

Hereditary Transthyretin-Related Amyloidosis Ongoing Observational Study: A Baseline Report of the First 3167 Participants
- PMID: 39458146
- PMCID: PMC11508262
- DOI: 10.3390/jcm13206197
Hereditary Transthyretin-Related Amyloidosis Ongoing Observational Study: A Baseline Report of the First 3167 Participants
Abstract
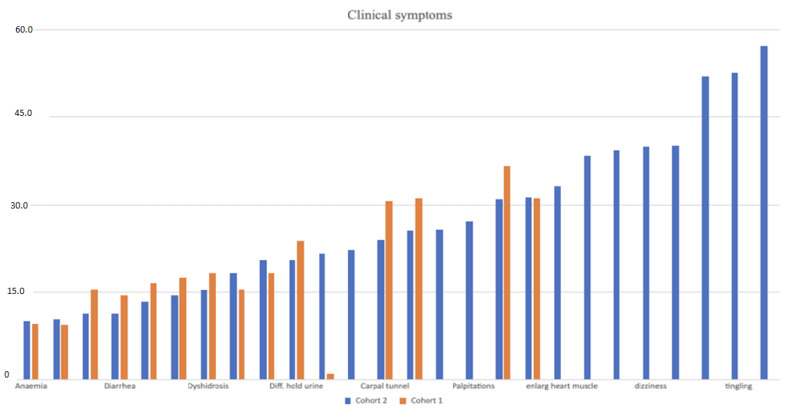
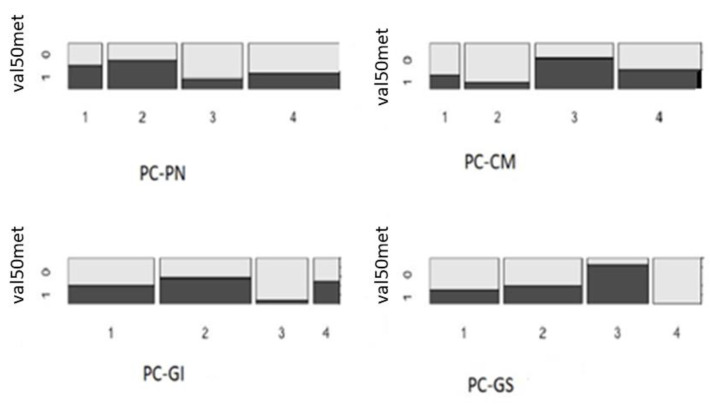
Background: Hereditary transthyretin-related amyloidosis is a clinically heterogeneous autosomal dominant disease caused by pathogenic variants in the TTR gene (hATTR amyloidosis). Objective: The current study describes the demographic, clinical, and genetic characteristics of patients with suspected hATTR amyloidosis. Methods: This study is part of the "Hereditary transthyretin-related amyloidosis and longitudinal monitoring of TTR-positive patients" (TRAMmoniTTR) study. This study included 3167 participants, along with their clinical details. Principal component (PC) analysis was used to analyze their clinical symptomatology. Next-generation sequencing of the TTR gene was performed and genotype-phenotype relationships were investigated. We compared the demographic and clinical characteristics using the principal components (PCs) and also compared participants with and without the TTR pathogenic variants. Results: We identified five main clinical phenotypes out of 22 single symptoms that explained 49% of the variation. The first two PCs referred to polyneuropathy and cardiomyopathy. We found significant differences between gender and PC-polyneuropathy and PC-cardiomyopathy, with male over-representation in the higher quantiles of PC-polyneuropathy and male under-representation in the lowest quantiles of PC-cardiomyopathy. We identified 92 participants with hATTR (3%), exhibiting 17 unique heterozygous TTR variants. The p.Val50Met variant was the most frequent. Furthermore, 503 participants (20%) were identified with ATTR and no relevant TTR variants (ATTRwt). We detected significant differences between the ATTRwt and hATTR groups, with male gender predominance in only the ATTRwt group and a positive family history of polyneuropathy and/or cardiomyopathy among the hATTR participants. Conclusions: The current clinical and genetic characterization of this cohort serves as a foundation for further longitudinal monitoring and assessment.
Keywords: Hereditary Transthyretin-Related Amyloidosis; TTR; hATTR amyloidosis.
Conflict of interest statement
S.R., L.M.P., A.M.B.-A., P.E., S.S. (Sabine Schröder), S.Z. (Susanne Zielke), V.B., J.K., G.W., F.R., S.S. (Snezana Skobalj), S.Z. (Susan Zielske), X.B., P.G., J.R., and P.B. are (or were) employees of Centogene at the time of the design and execution of this study. K.H. received financial reimbursement for consulting, advisory board activities, speaker fees, and/or contributions to congresses and travel support to attend scientific meetings by Akcea Therapeuticals Inc., Alnylam Pharmaceuticals Inc., Amicus, AstraZeneca, GSK, Hormosan, Takeda Pharmaceutical Inc., Pfizer Pharmaceuticals Inc., Swedish Orphan Biovitrum Inc., and ViiV Healthcare GmbH. K.H. further received research funding by the foundation Charité (BIH clinical fellow), Alnylam Pharmaceuticals Inc., and Pfizer Pharmaceuticals. All authors declare no conflicts of interest pertaining to their contribution to this work.
Figures
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
