

Molecular Dynamics (MD) Simulations Provide Insights into the Activation Mechanisms of 5-HT2A Receptors
- PMID: 39459303
- PMCID: PMC11510212
- DOI: 10.3390/molecules29204935
Molecular Dynamics (MD) Simulations Provide Insights into the Activation Mechanisms of 5-HT2A Receptors
Abstract
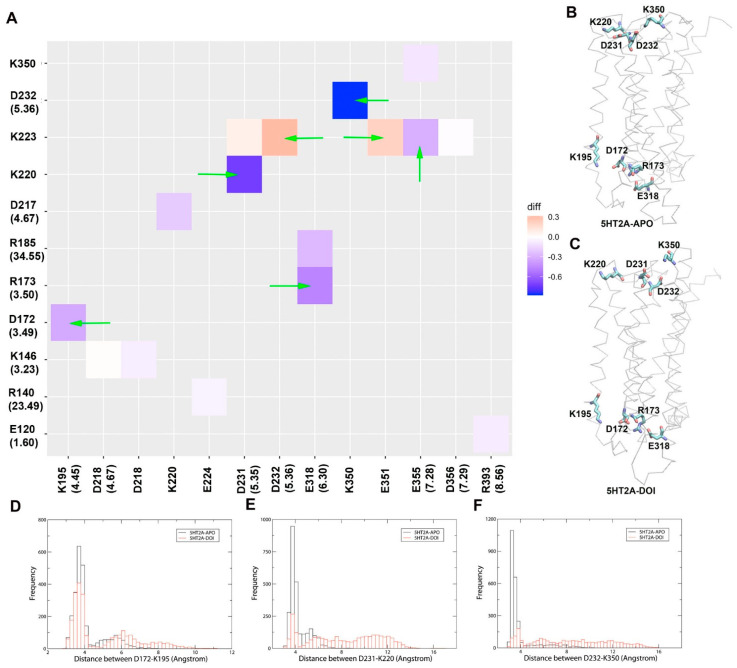
Recent breakthroughs in the determination of atomic resolution 3-D cryo-electron microscopy structures of membrane proteins present an unprecedented opportunity for drug discovery. Structure-based drug discovery utilizing in silico methods enables the study of dynamic connectivity of stable conformations induced by the drug in achieving its effect. With the ever-expanding computational power, simulations of this type reveal protein dynamics in the nano-, micro-, and even millisecond time scales. In the present study, aiming to characterize the protein dynamics of the 5HT2A receptor stimulated by ligands (agonist/antagonist), we performed 1 µs MD simulations on 5HT2A/DOI (agonist), 5HT2A/GSK215083 (antagonist), and 5HT2A (APO, no ligand) systems. The crystal structure of 5HT2A/zotepine (antagonist) (PDB: 6A94) was used to set up the simulation systems in a lipid bilayer environment. We found the monitoring of the ionic lock residue pair (R3.50-E6.30) of 5HT2A in MD simulations to be a good approximation of the effects of agonists (ionic lock breakage) or antagonists (ionic lock formation) on receptor activation. We further performed analyses of the MD trajectories, including Principal Component Analysis (PCA), hydrogen bond, salt bridge, and hydrophobic interaction network analyses, and correlation between residues to identify key elements of receptor activation. Our results suggest that in order to trigger receptor activation, DOI must interact with 5HT2A through residues V5.39, G5.42, S5.43, and S5.46 on TM5, inducing significant conformational changes in the backbone angles of G5.42 and S5.43. DOI also interacted with residues W6.48 (toggle switch) and F6.51 on TM6, causing major conformational shifts in the backbone angles of F6.44 and V6.45. These structural changes were transmitted to the intracellular ends of TM5, TM6, and ICL3, resulting in the breaking of the ionic lock and subsequent G protein activation. The studies could be helpful in future design of selective agonists/antagonists for various serotonin receptors (5HT1A, 5HT2A, 5HT2B, 5HT2C, and 5HT7) involved in detrimental disorders, such as addiction and schizophrenia.
Keywords: 5HT2A receptor; G-protein-coupled receptor (GPCR); MD simulations; conformational changes; protein−ligand interactions; receptor activation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures








References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
