

Comprehensive reanalysis for CNVs in ES data from unsolved rare disease cases results in new diagnoses
- PMID: 39461972
- PMCID: PMC11513043
- DOI: 10.1038/s41525-024-00436-6
Comprehensive reanalysis for CNVs in ES data from unsolved rare disease cases results in new diagnoses
Abstract
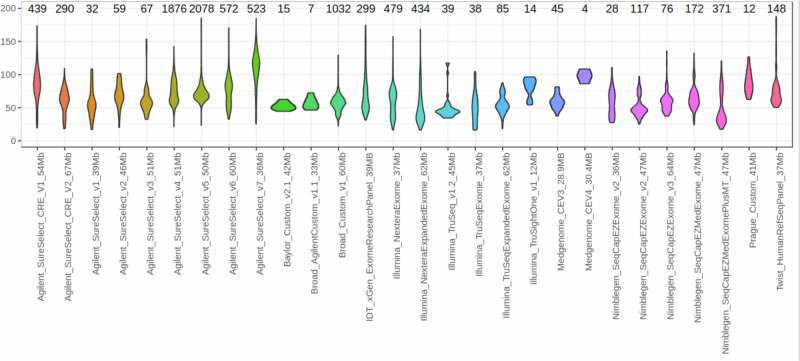
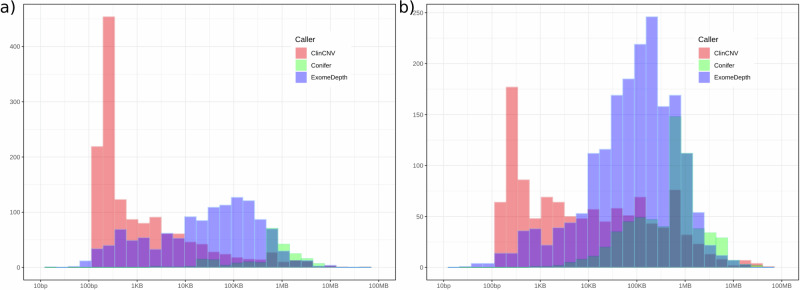
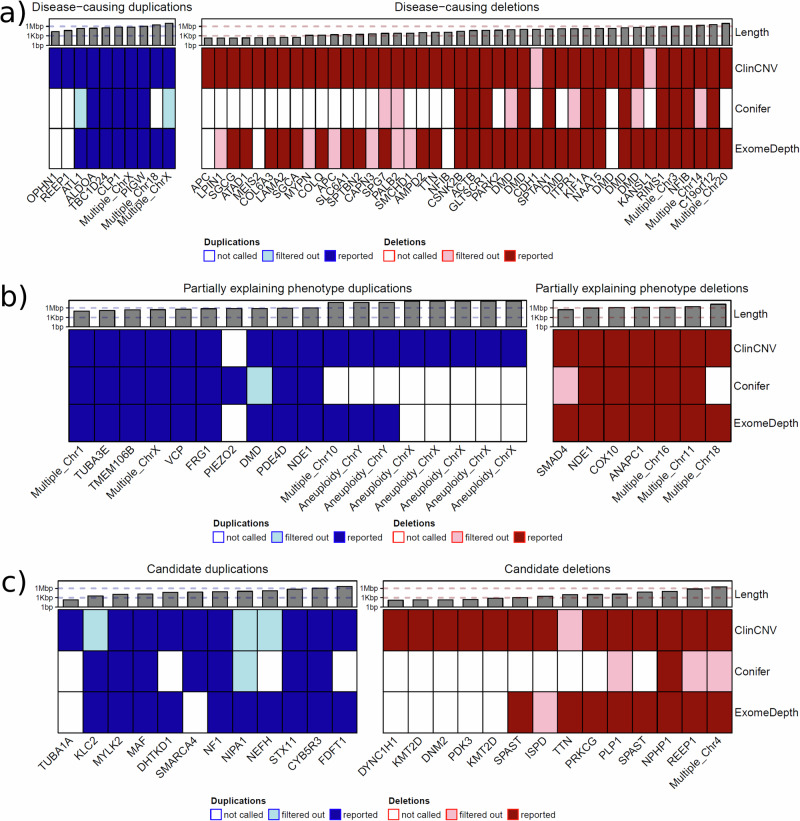
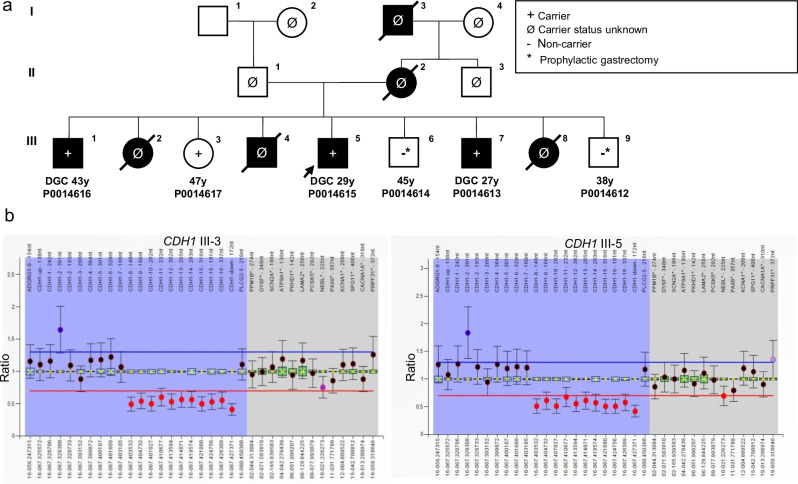
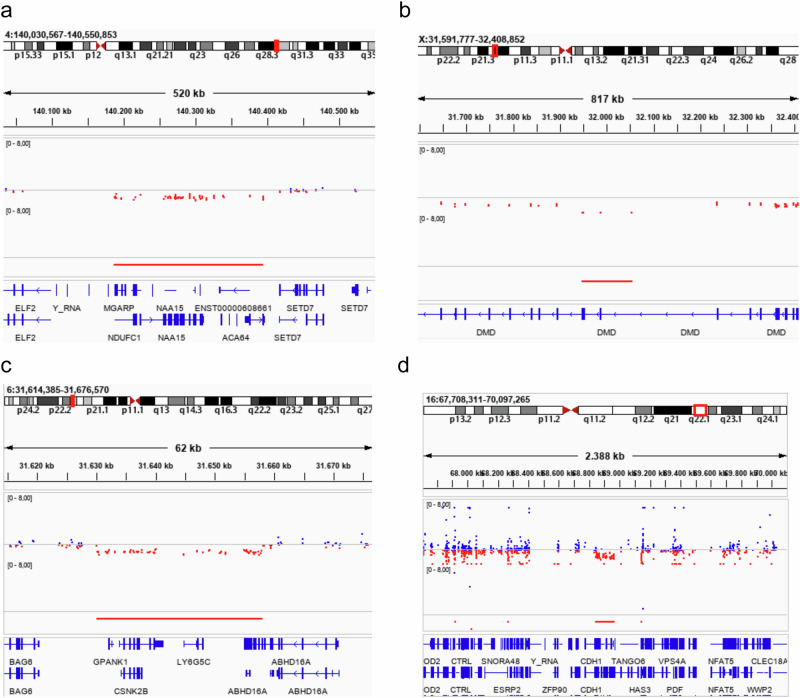
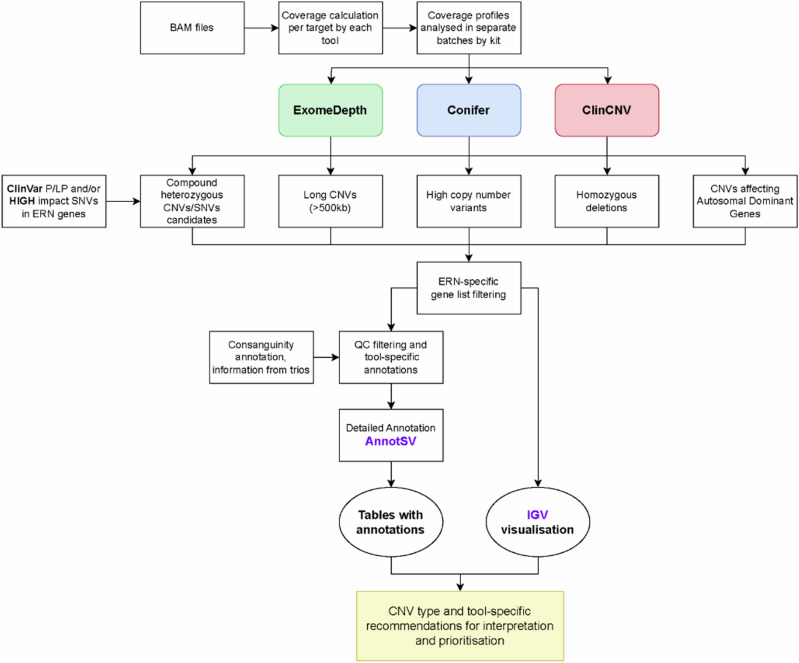
We report the results of a comprehensive copy number variant (CNV) reanalysis of 9171 exome sequencing datasets from 5757 families affected by a rare disease (RD). The data reanalysed was extremely heterogeneous, having been generated using 28 different enrichment kits by 42 different research groups across Europe partnering in the Solve-RD project. Each research group had previously undertaken their own analysis of the data but failed to identify disease-causing variants. We applied three CNV calling algorithms to maximise sensitivity, and rare CNVs overlapping genes of interest, provided by four partner European Reference Networks, were taken forward for interpretation by clinical experts. This reanalysis has resulted in a molecular diagnosis being provided to 51 families in this sample, with ClinCNV performing the best of the three algorithms. We also identified partially explanatory pathogenic CNVs in a further 34 individuals. This work illustrates the value of reanalysing ES cold cases for CNVs.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- European Commission. EU Research on Rare Diseaseshttps://research-and-innovation.ec.europa.eu/research-area/health/rare-d... (2023).
-
- Poplin, R. et al. Scaling accurate genetic variant discovery to tens of thousands of samples. Preprint at bioRxiv10.1101/201178 (2018).
LinkOut - more resources
Full Text Sources
Miscellaneous
