

This is a preprint.
Expanding and improving analyses of nucleotide recoding RNA-seq experiments with the EZbakR suite
- PMID: 39463977
- PMCID: PMC11507695
- DOI: 10.1101/2024.10.14.617411
Expanding and improving analyses of nucleotide recoding RNA-seq experiments with the EZbakR suite
Update in
-
Expanding and improving analyses of nucleotide recoding RNA-seq experiments with the EZbakR suite.PLoS Comput Biol. 2025 Jul 3;21(7):e1013179. doi: 10.1371/journal.pcbi.1013179. eCollection 2025 Jul. PLoS Comput Biol. 2025. PMID: 40609070 Free PMC article.
Abstract
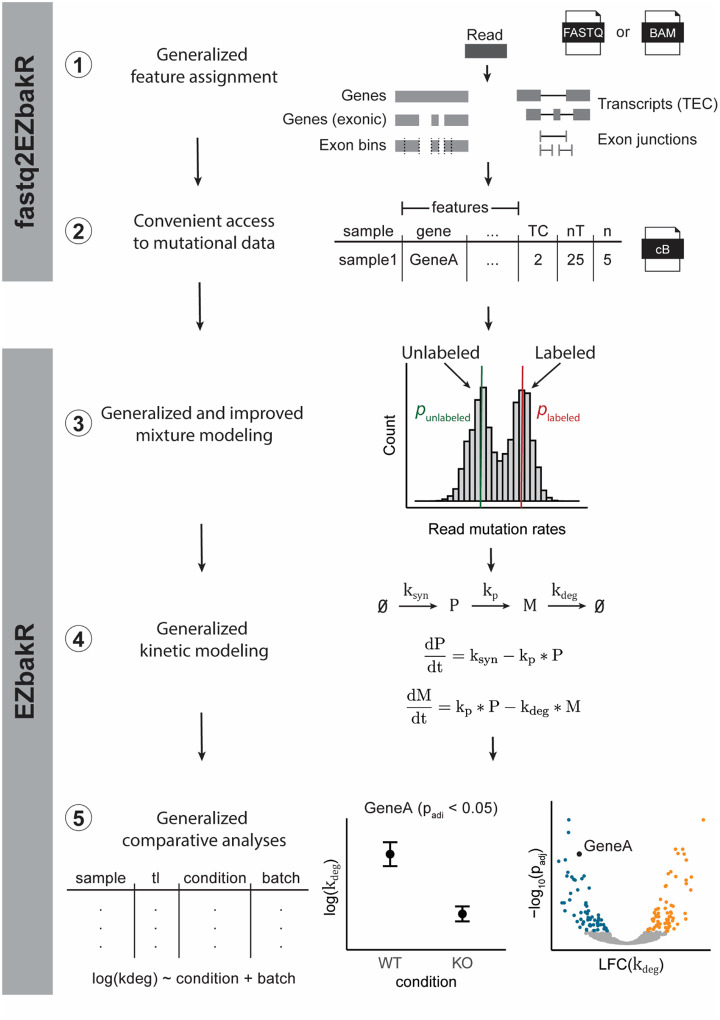
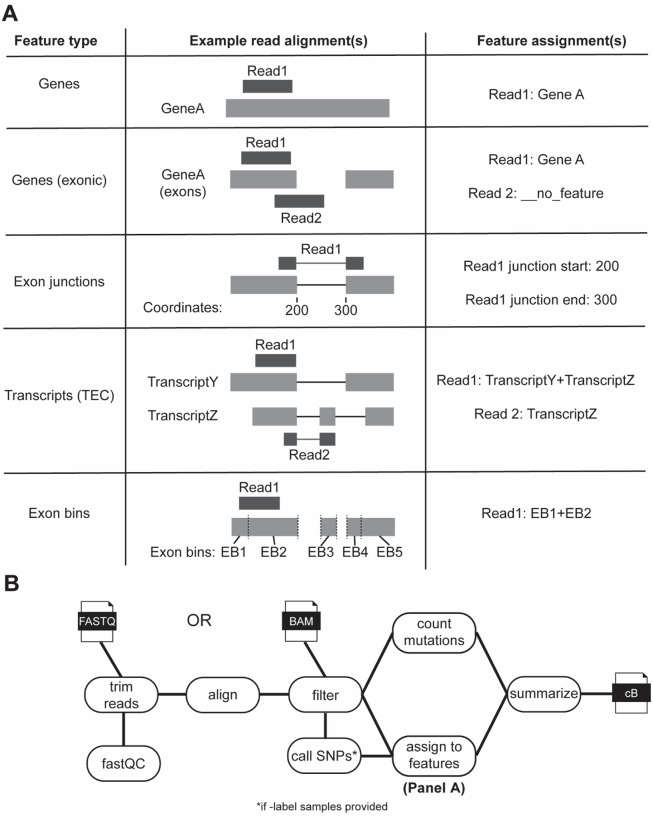
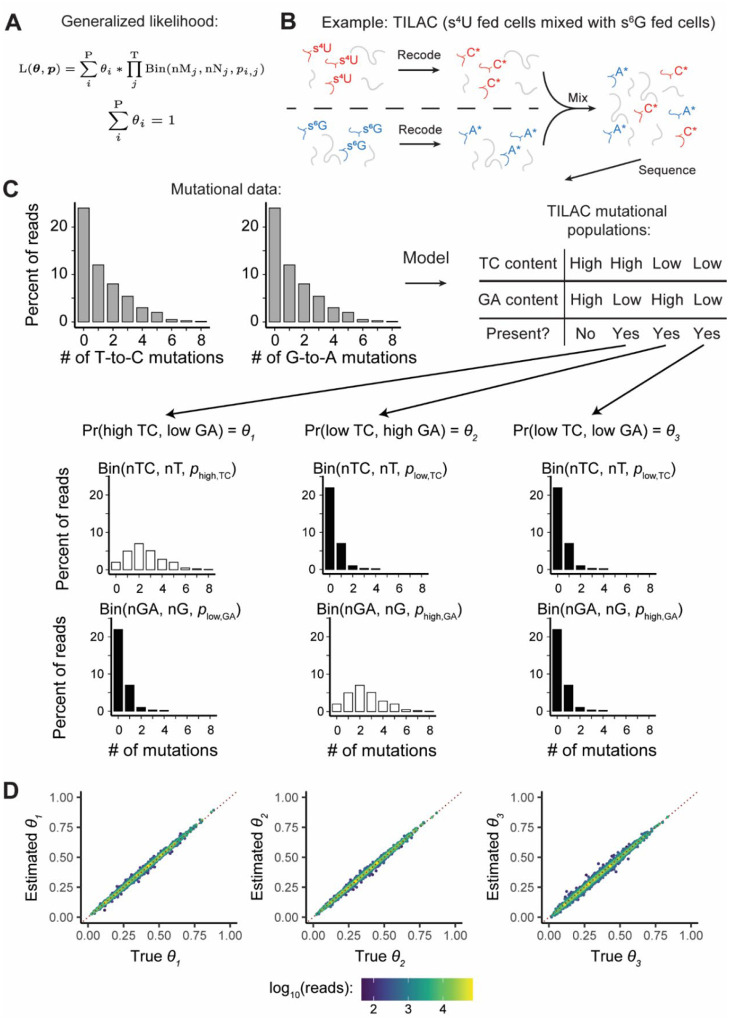
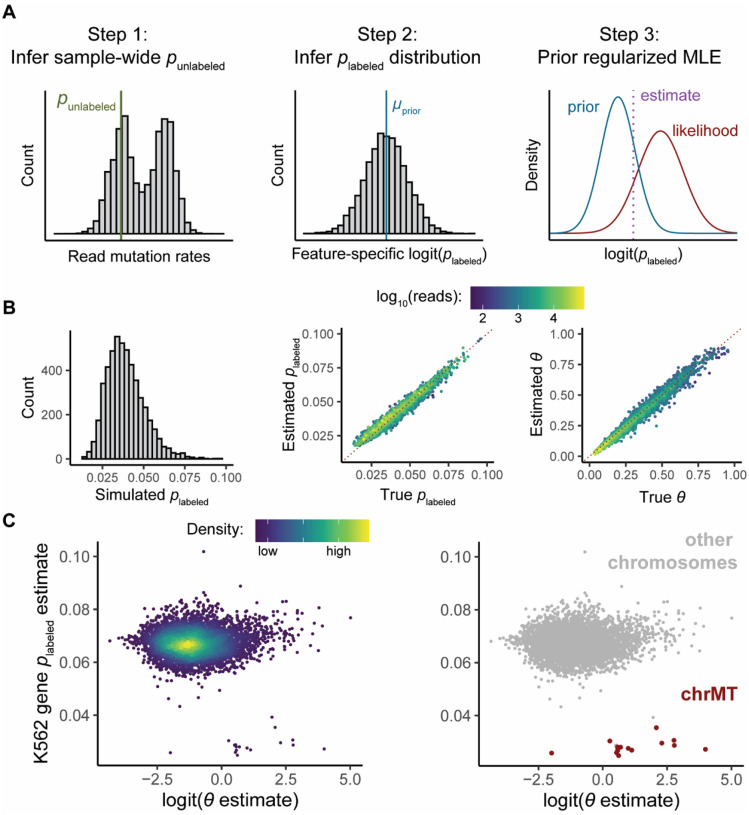
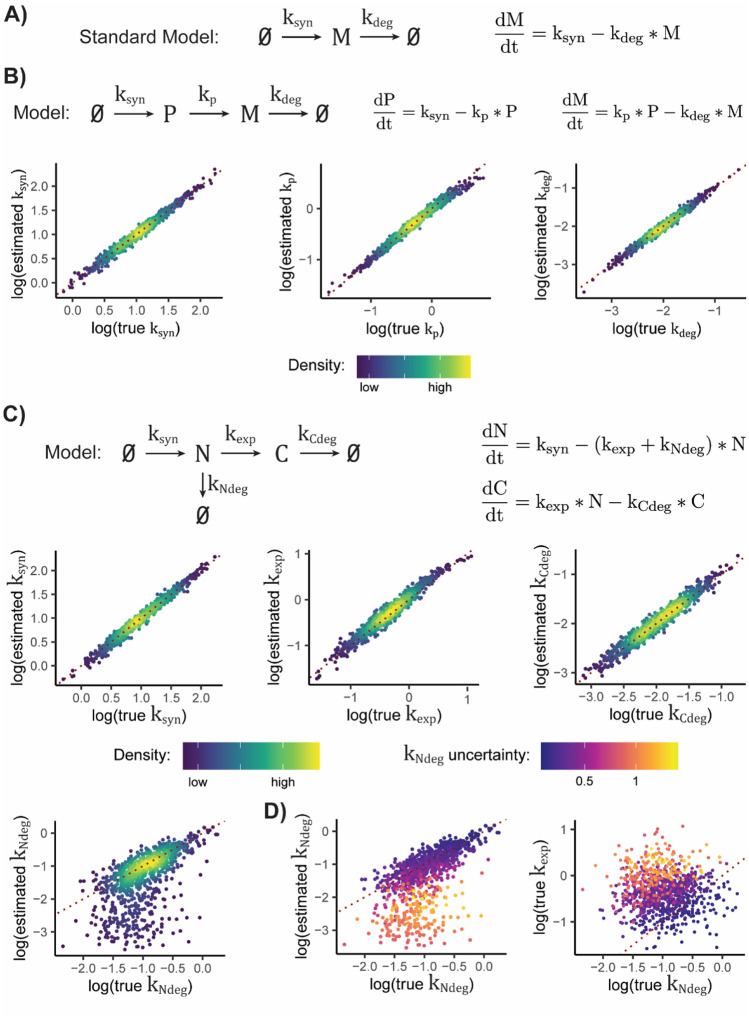
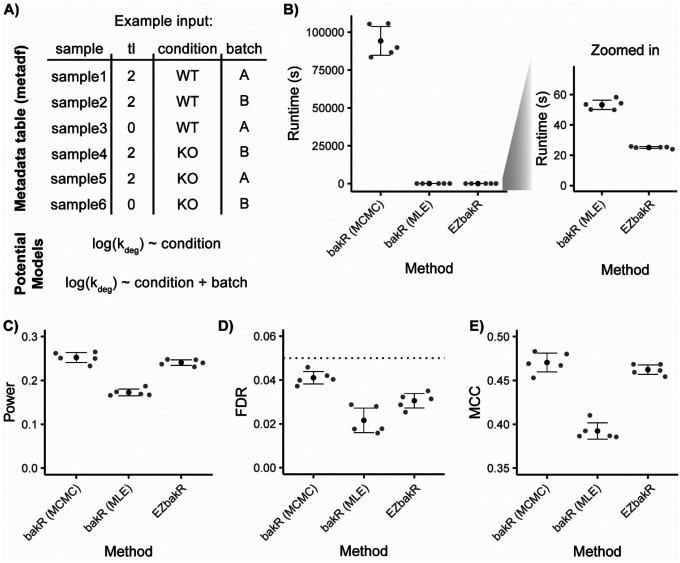
Nucleotide recoding RNA sequencing methods (NR-seq; TimeLapse-seq, SLAM-seq, TUC-seq, etc.) are powerful approaches for assaying transcript population dynamics. In addition, these methods have been extended to probe a host of regulated steps in the RNA life cycle. Current bioinformatic tools significantly constrain analyses of NR-seq data. To address this limitation, we developed EZbakR, an R package to facilitate a more comprehensive set of NR-seq analyses, and fastq2EZbakR, a Snakemake pipeline for flexible preprocessing of NR-seq datasets, collectively referred to as the EZbakR suite. Together, these tools generalize many aspects of the NR-seq analysis workflow. The fastq2EZbakR pipeline can assign reads to a diverse set of genomic features (e.g., genes, exons, splice junctions, etc.), and EZbakR can perform analyses on any combination of these features. EZbakR extends standard NR-seq mutational modeling to support multi-label analyses (e.g., s4U and s6G dual labeling), and implements an improved hierarchical model to better account for transcript-to-transcript variance in metabolic label incorporation. EZbakR also generalizes dynamical systems modeling of NR-seq data to support analyses of premature mRNA processing and flow between subcellular compartments. Finally, EZbakR implements flexible and well-powered comparative analyses of all estimated parameters via design matrix-specified generalized linear modeling. The EZbakR suite will thus allow researchers to make full, effective use of NR-seq data.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources