

This is a preprint.
Non-lytic spread of poliovirus requires the nonstructural protein 3CD
- PMID: 39464037
- PMCID: PMC11507938
- DOI: 10.1101/2024.10.18.619132
Non-lytic spread of poliovirus requires the nonstructural protein 3CD
Update in
-
Non-lytic spread of poliovirus requires the nonstructural protein 3CD.mBio. 2025 Jan 8;16(1):e0327624. doi: 10.1128/mbio.03276-24. Epub 2024 Dec 12. mBio. 2025. PMID: 39665531 Free PMC article.
Abstract
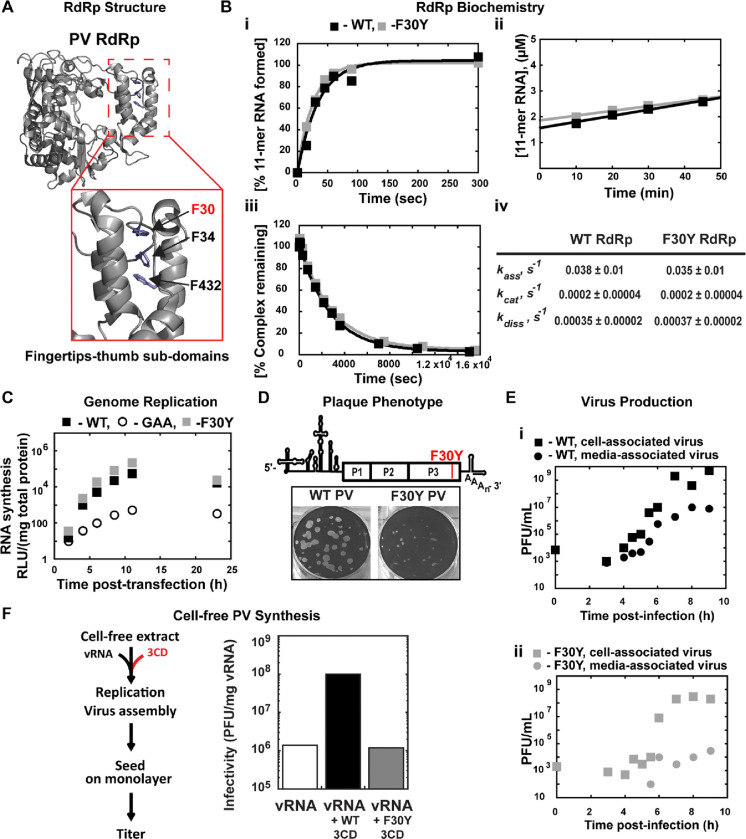
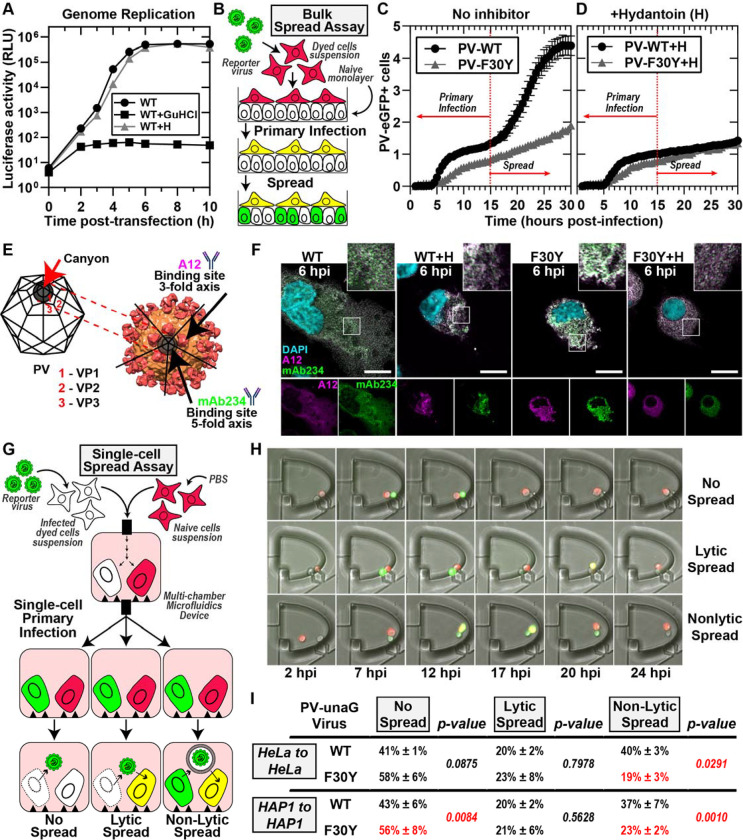
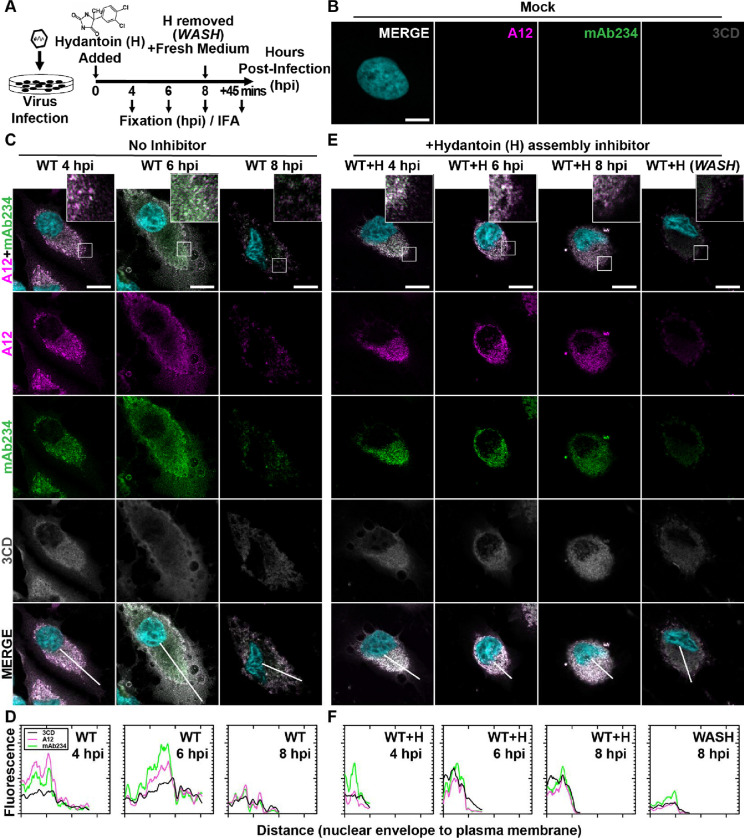
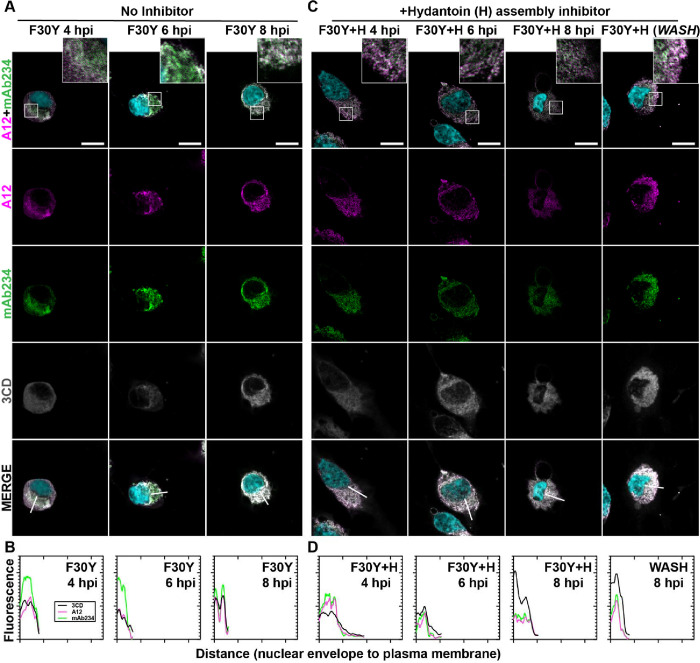
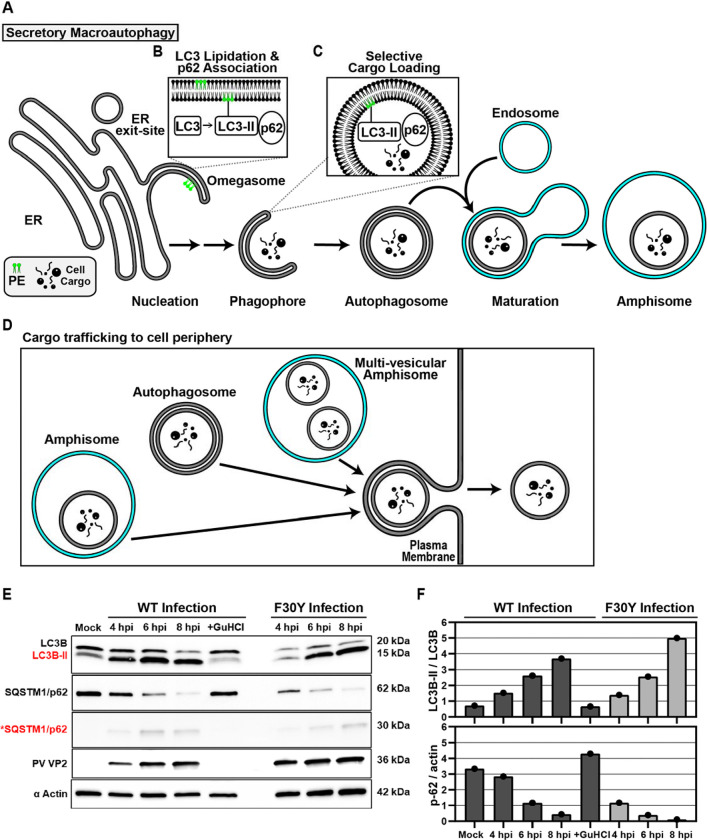
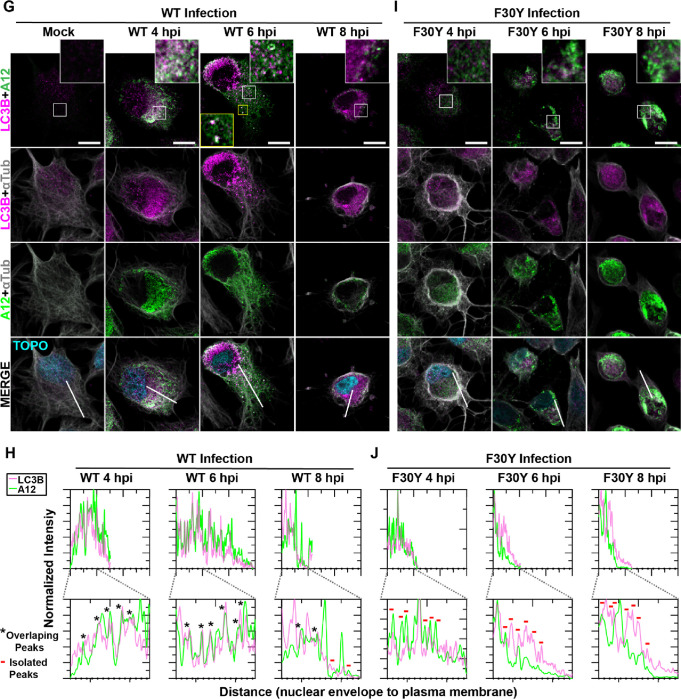
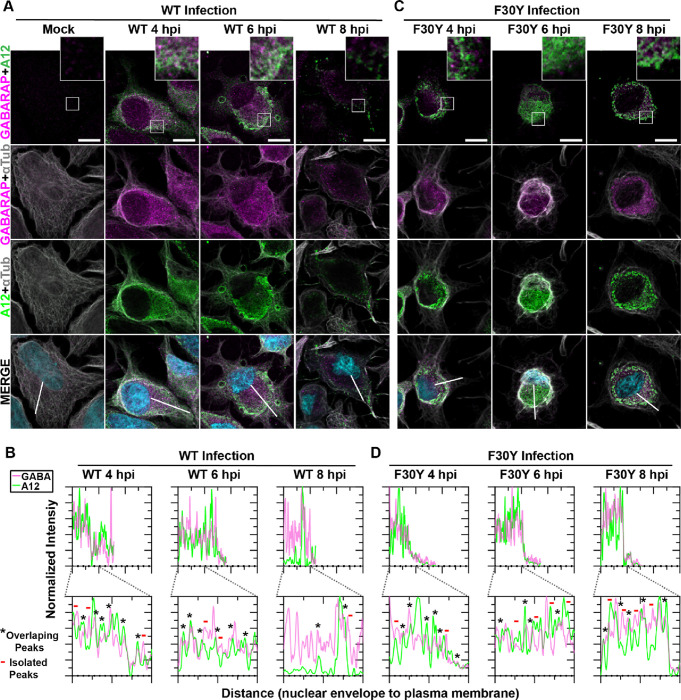
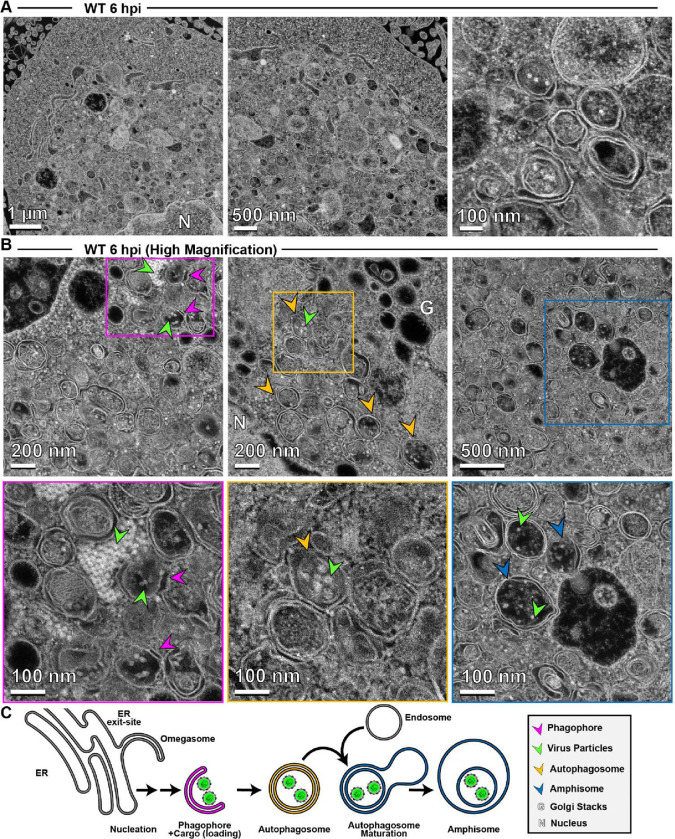
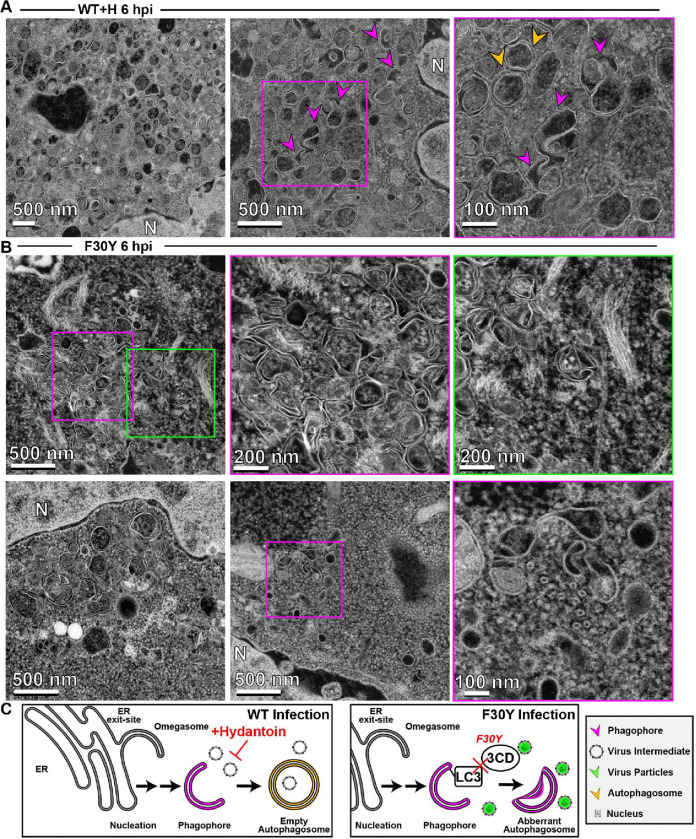
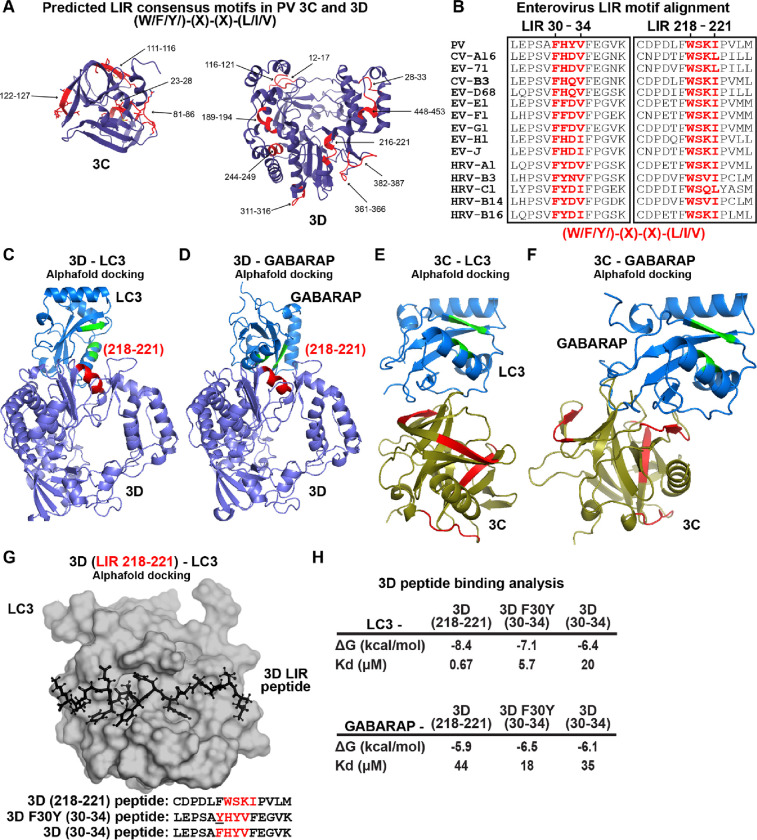
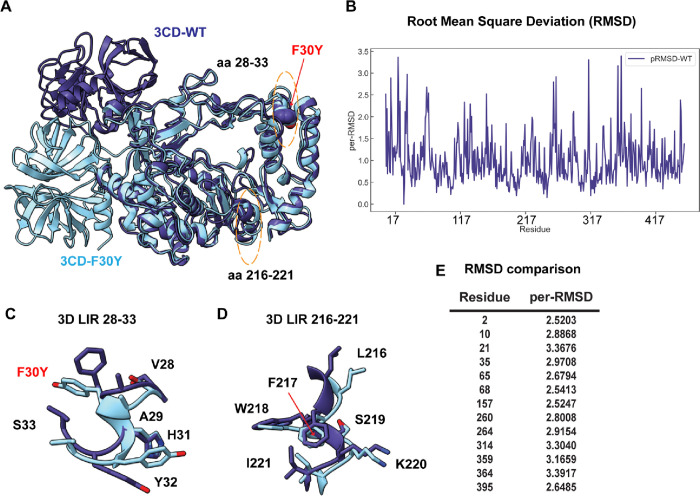
Non-enveloped viruses like poliovirus (PV) have evolved the capacity to spread by non-lytic mechanisms. For PV, this mechanism exploits the host secretory autophagy pathway. Virions are selectively incorporated into autophagosomes, double-membrane vesicles that travel to the plasma membrane, fuse, and release single-membrane vesicles containing virions. Loading of cellular cargo into autophagosomes relies on direct or indirect interactions with microtubule-associated protein 1B-light chain 3 (LC3) that are mediated by motifs referred to as LC3-interaction regions (LIRs). We have identified a PV mutant with a severe defect in non-lytic spread. An F-to-Y substitution in a putative LIR of the nonstructural protein 3CD prevented virion incorporation into LC3-positive autophagosomes and virion trafficking to the plasma membrane for release. Using high-angle annular dark-field scanning transmission electron microscopy to monitor PV-induced autophagosome biogenesis, for the first time, we show that virus-induced autophagic signals yield normal autophagosomes, even in the absence of virions. The F-to-Y derivative of PV 3CD was unable to support normal autophagosome biogenesis. Together, these studies make a compelling case for a direct role of a viral nonstructural protein in the formation and loading of the vesicular carriers used for non-lytic spread that may depend on the proper structure, accessibility, and/or dynamics of its LIR. The studies of PV 3CD protein reported here will hopefully provoke a more deliberate look at the presence and function of LIR motifs in viral proteins of viruses known to use autophagy as the basis for non-lytic spread.
Figures
References
-
- Andino R, Kirkegaard K, Macadam A, Racaniello VR, Rosenfeld AB. 2023. The Picornaviridae Family: Knowledge Gaps, Animal Models, Countermeasures, and Prototype Pathogens. J Infect Dis 228:S427–S445. - PubMed
-
- Chumakov K, Ehrenfeld E, Agol VI, Wimmer E. 2021. Polio eradication at the crossroads. Lancet Glob Heal 9:e1172–e1175. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources