

Fenebrutinib, a Bruton's tyrosine kinase inhibitor, blocks distinct human microglial signaling pathways
- PMID: 39465429
- PMCID: PMC11514909
- DOI: 10.1186/s12974-024-03267-5
Fenebrutinib, a Bruton's tyrosine kinase inhibitor, blocks distinct human microglial signaling pathways
Abstract
Background: Bruton's tyrosine kinase (BTK) is an intracellular signaling enzyme that regulates B-lymphocyte and myeloid cell functions. Due to its involvement in both innate and adaptive immune compartments, BTK inhibitors have emerged as a therapeutic option in autoimmune disorders such as multiple sclerosis (MS). Brain-penetrant, small-molecule BTK inhibitors may also address compartmentalized neuroinflammation, which is proposed to underlie MS disease progression. BTK is expressed by microglia, which are the resident innate immune cells of the brain; however, the precise roles of microglial BTK and impact of BTK inhibitors on microglial functions are still being elucidated. Research on the effects of BTK inhibitors has been limited to rodent disease models. This is the first study reporting effects in human microglia.
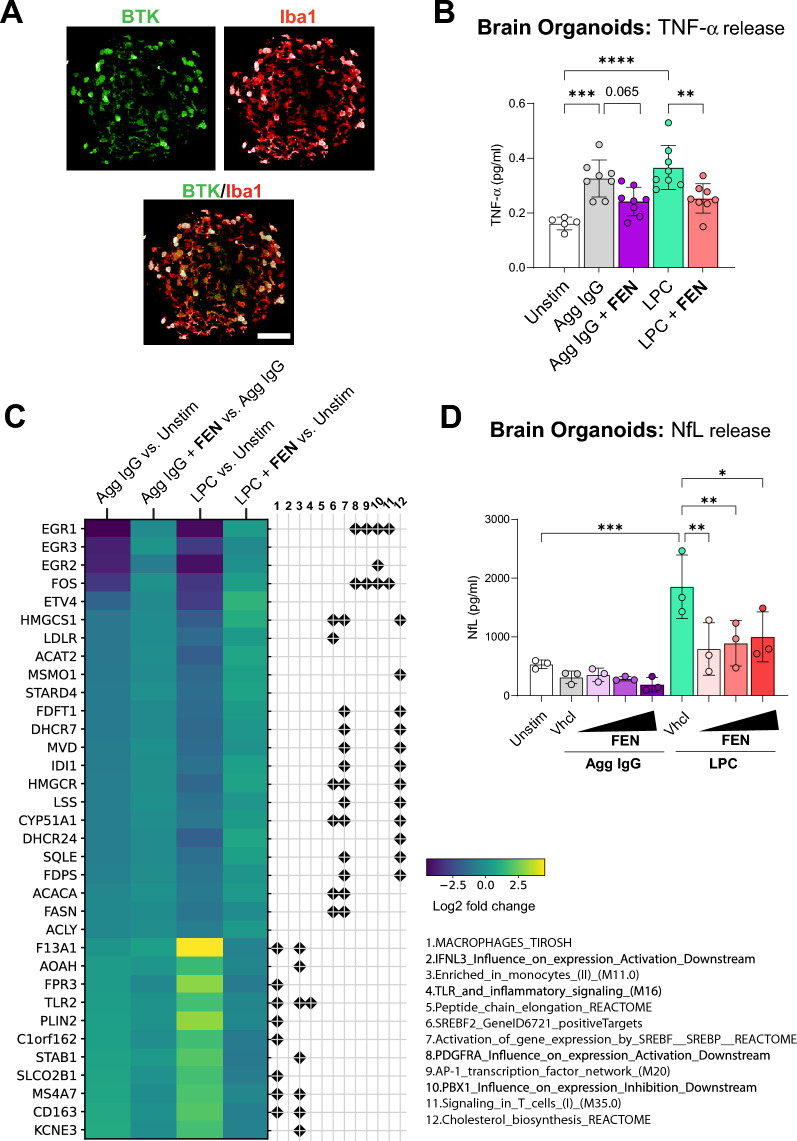
Methods: Here we characterize the pharmacological and functional properties of fenebrutinib, a potent, highly selective, noncovalent, reversible, brain-penetrant BTK inhibitor, in human microglia and complex human brain cell systems, including brain organoids.
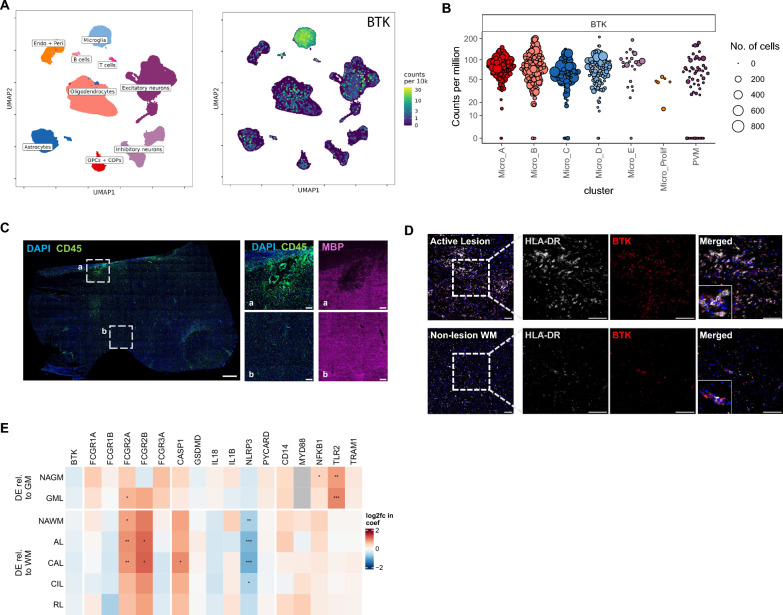
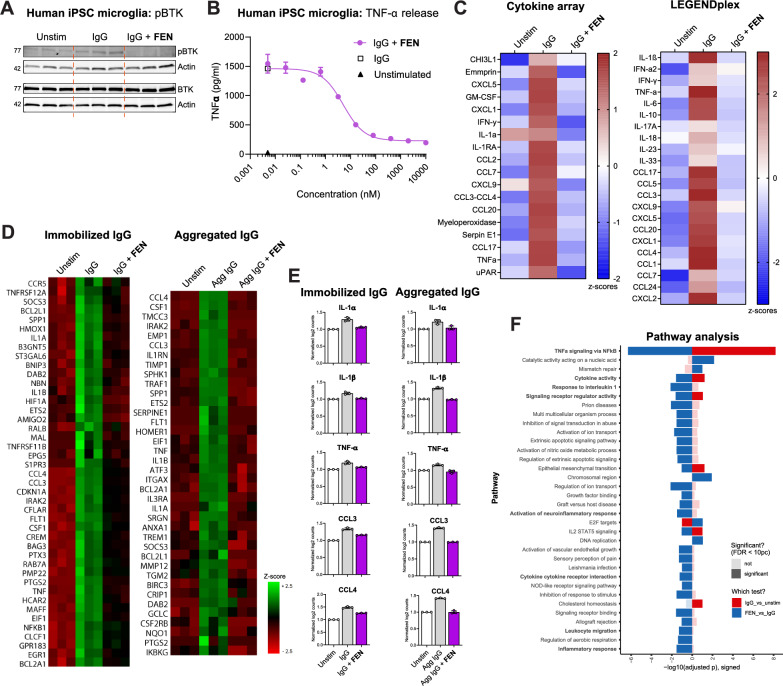
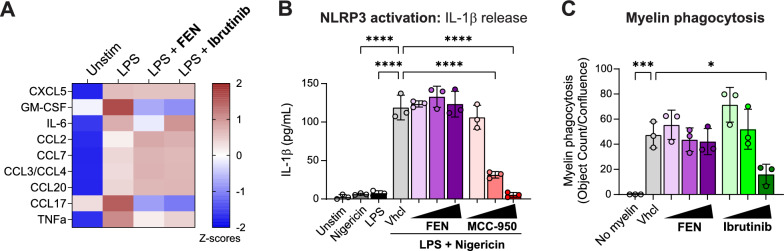
Results: We find that fenebrutinib blocks the deleterious effects of microglial Fc gamma receptor (FcγR) activation, including cytokine and chemokine release, microglial clustering and neurite damage in diverse human brain cell systems. Gene expression analyses identified pathways linked to inflammation, matrix metalloproteinase production and cholesterol metabolism that were modulated by fenebrutinib treatment. In contrast, fenebrutinib had no significant impact on human microglial pathways linked to Toll-like receptor 4 (TLR4) and NACHT, LRR and PYD domains-containing protein 3 (NLRP3) signaling or myelin phagocytosis.
Conclusions: Our study enhances the understanding of BTK functions in human microglial signaling that are relevant to MS pathogenesis and suggests that fenebrutinib could attenuate detrimental microglial activity associated with FcγR activation in people with MS.
Keywords: BTK; Fenebrutinib; Microglia; Multiple sclerosis; Neuroinflammation; Organoids.
© 2024. The Author(s).
Conflict of interest statement
JL, SL, ME, WM, RS, JD, TS, LC and JK were employees and shareholders of F. Hoffmann-La Roche Ltd during the time that this work was completed. YS and CH were employees of Genentech, Inc., and shareholders of F. Hoffmann-La Roche Ltd during the time this work was completed. VWY has research contracts with Genentech, Inc. and Novartis. He has received speaker honoraria from Biogen Idec, EMD Serono (Merck), Novartis, F. Hoffmann-La Roche Ltd, Sanofi Genzyme and Teva Canada. He is the recipient of unrestricted educational grants from Biogen Idec, EMD Serono (Merck), Novartis, F. Hoffmann-La Roche Ltd, Sanofi Genzyme and Teva Canada to support educational activities of the Alberta MS Network and Americas School of Neuroimmunology of the International Society of Neuroimmunology, which he directs.
Figures


References
-
- Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14:219–32. - PubMed
-
- Jongstra-Bilen J, Puig Cano A, Hasija M, Xiao H, Smith CI, Cybulsky MI. Dual functions of Bruton’s tyrosine kinase and Tec kinase during Fcgamma receptor-induced signaling and phagocytosis. J Immunol. 2008;181:288–98. - PubMed
-
- Ellmeier W, Abramova A, Schebesta A. Tec family kinases: regulation of FcεRI-mediated mast-cell activation. FEBS J. 2011;278:1990–2000. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
