

Phosphorylation of P-stalk proteins defines the ribosomal state for interaction with auxiliary protein factors
- PMID: 39468350
- PMCID: PMC11624264
- DOI: 10.1038/s44319-024-00297-1
Phosphorylation of P-stalk proteins defines the ribosomal state for interaction with auxiliary protein factors
Abstract
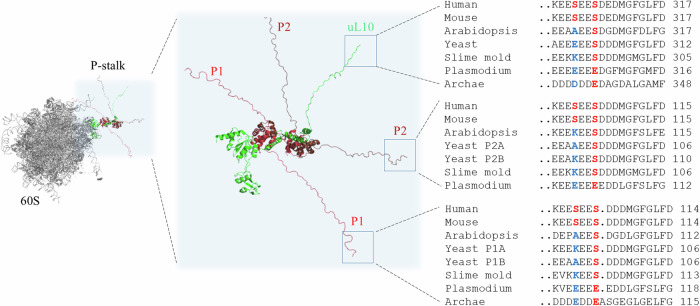
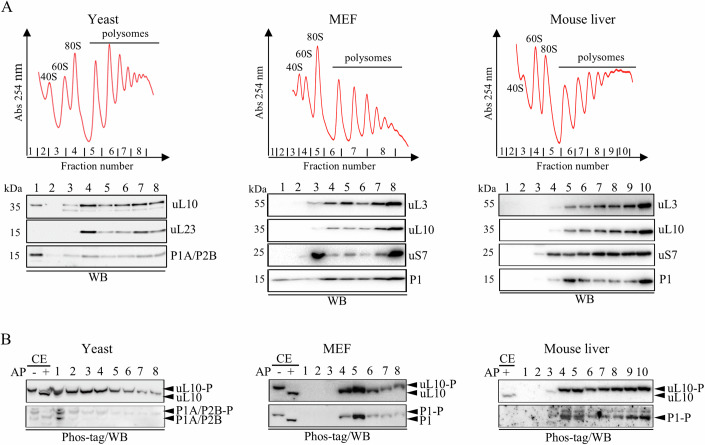
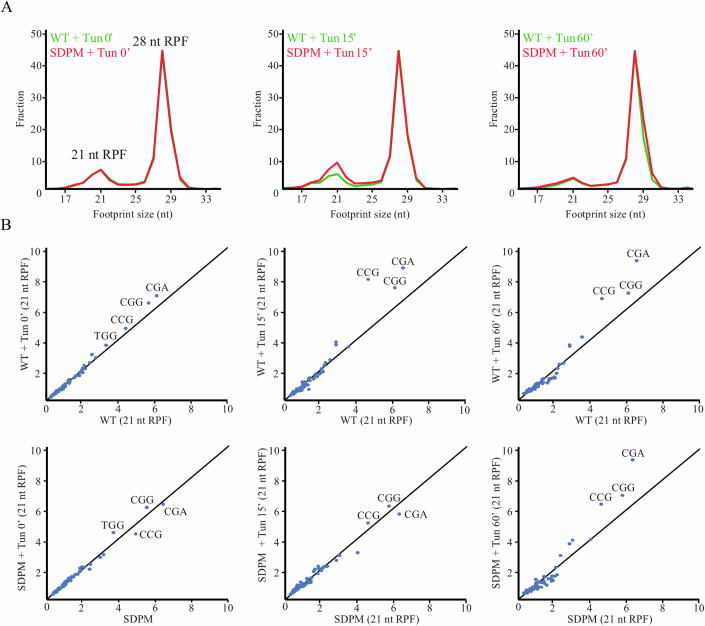
Ribosomal action is facilitated by the orchestrated work of trans-acting factors and ribosomal elements, which are subject to regulatory events, often involving phosphorylation. One such element is the ribosomal P-stalk, which plays a dual function: it activates translational GTPases, which support basic ribosomal functions, and interacts with the Gcn2 kinase, linking the ribosomes to the ISR pathway. We show that P-stalk proteins, which form a pentamer, exist in the cell exclusively in a phosphorylated state at five C-terminal domains (CTDs), ensuring optimal translation (speed and accuracy) and may play a role in the timely regulation of the Gcn2-dependent stress response. Phosphorylation of the CTD induces a structural transition from a collapsed to a coil-like structure, and the CTD gains conformational freedom, allowing specific but transient binding to various protein partners, optimizing the ribosome action. The report reveals a unique feature of the P-stalk proteins, indicating that, unlike most ribosomal proteins, which are regulated by phosphorylation in an on/off manner, the P-stalk proteins exist in a constantly phosphorylated state, which optimizes their interaction with auxiliary factors.
Keywords: Gcn2 Kinase; Phosphorylation; Ribosomal Proteins; Ribosomal Stalk; Ribosome.
© 2024. The Author(s).
Conflict of interest statement
Disclosure and competing interests statement. The authors declare no competing interests.
Figures












References
-
- Bah A, Vernon RM, Siddiqui Z, Krzeminski M, Muhandiram R, Zhao C, Sonenberg N, Kay LE, Forman-Kay JD (2015) Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 519:106–109 - PubMed
-
- Belin S, Hacot S, Daudignon L, Therizols G, Pourpe S, Mertani HC, Rosa-Calatrava M, Diaz JJ (2010) Purification of ribosomes from human cell lines. Curr Protoc Cell Biol 49:3–40 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
