

Longitudinal transcriptional changes reveal genes from the natural killer cell-mediated cytotoxicity pathway as critical players underlying COVID-19 progression
- PMID: 39470726
- PMCID: PMC11521369
- DOI: 10.7554/eLife.94242
Longitudinal transcriptional changes reveal genes from the natural killer cell-mediated cytotoxicity pathway as critical players underlying COVID-19 progression
Abstract
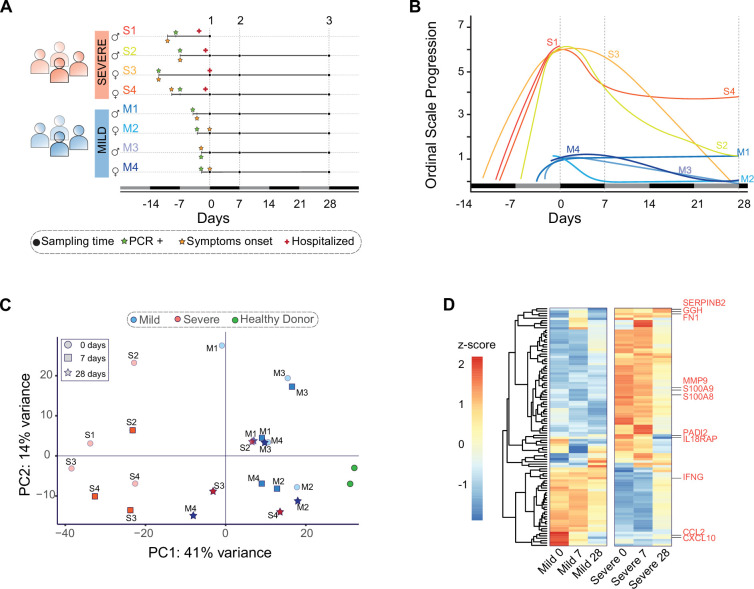
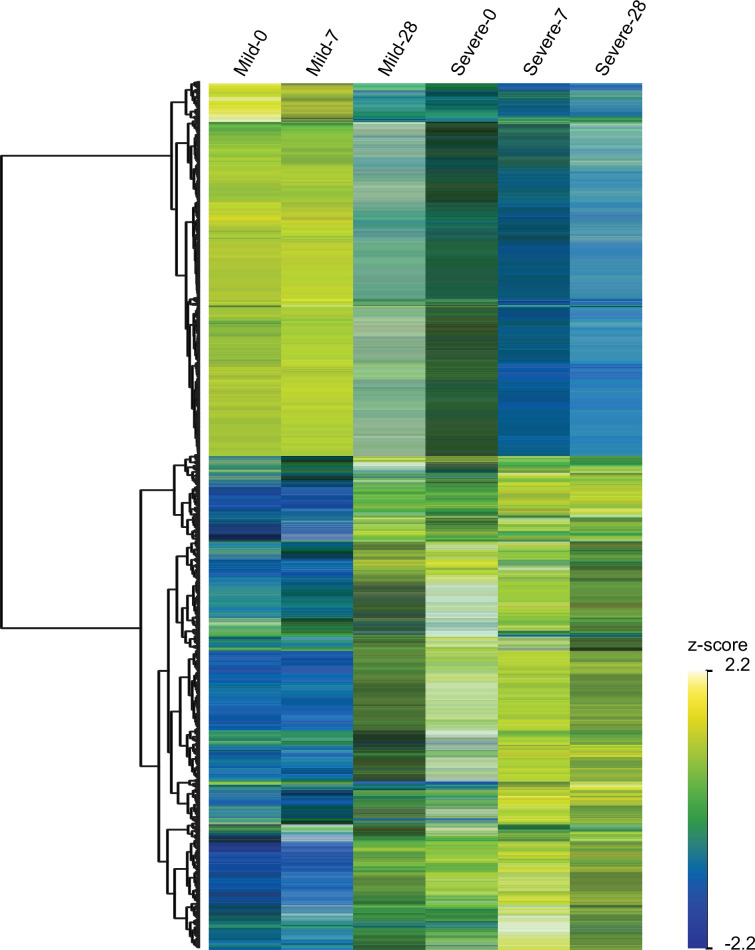
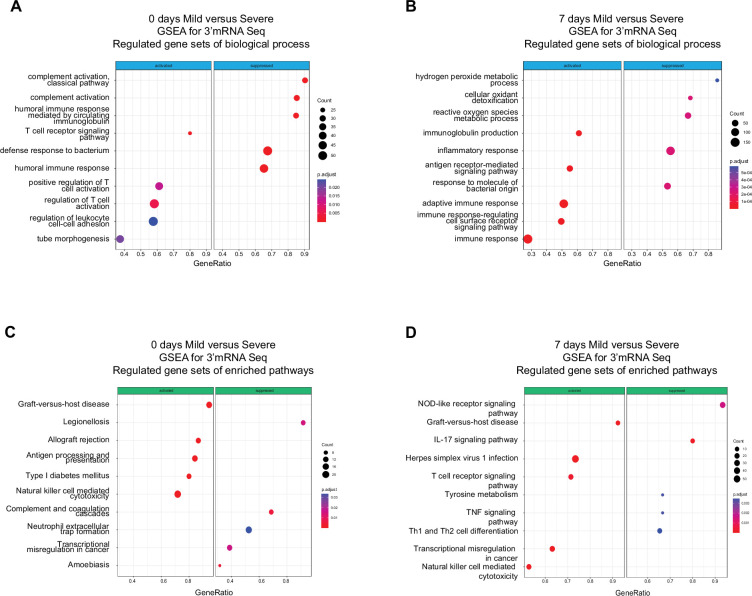
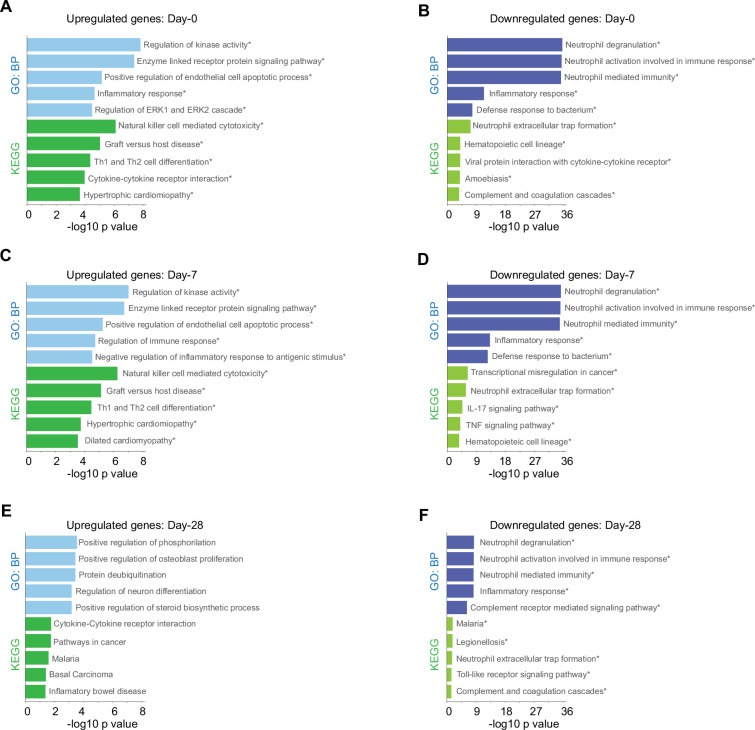
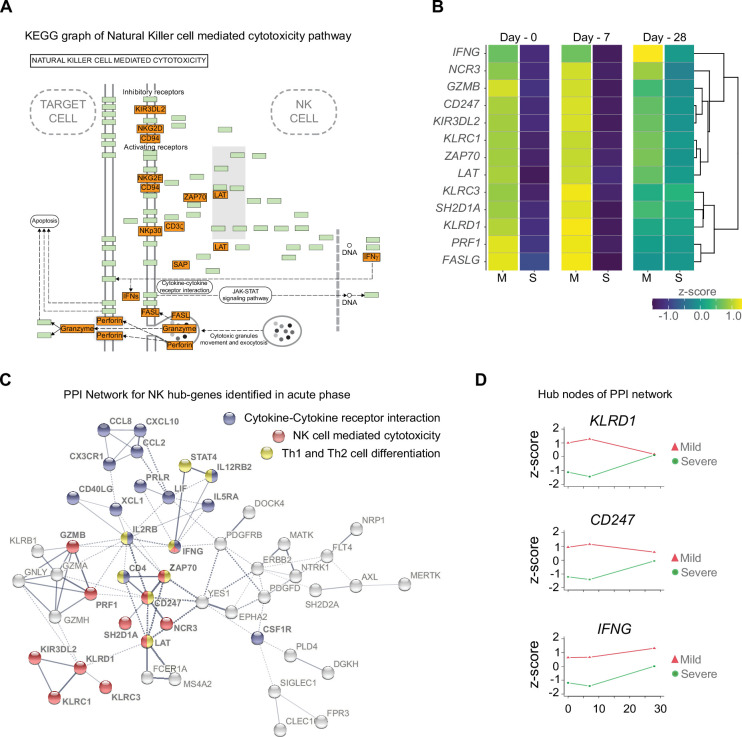
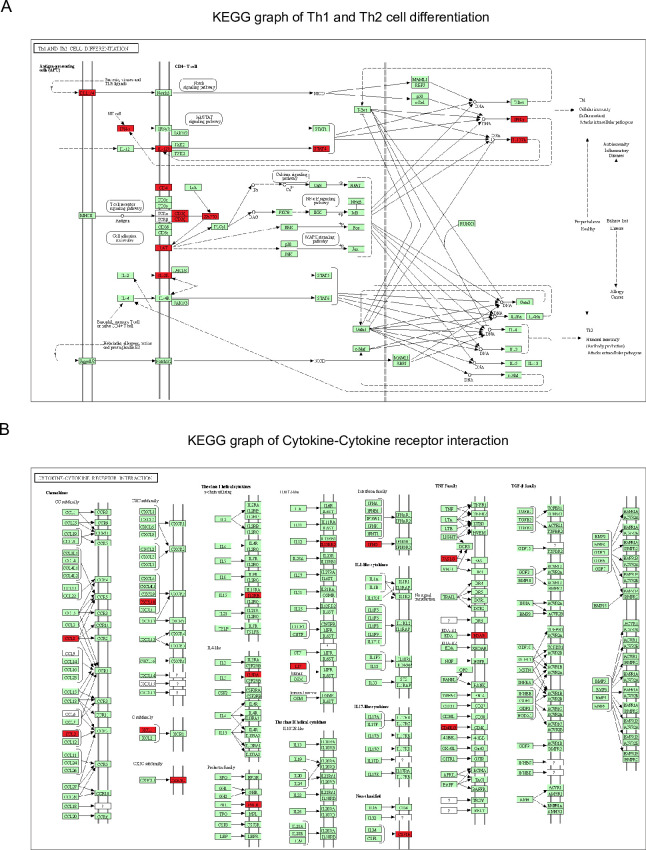
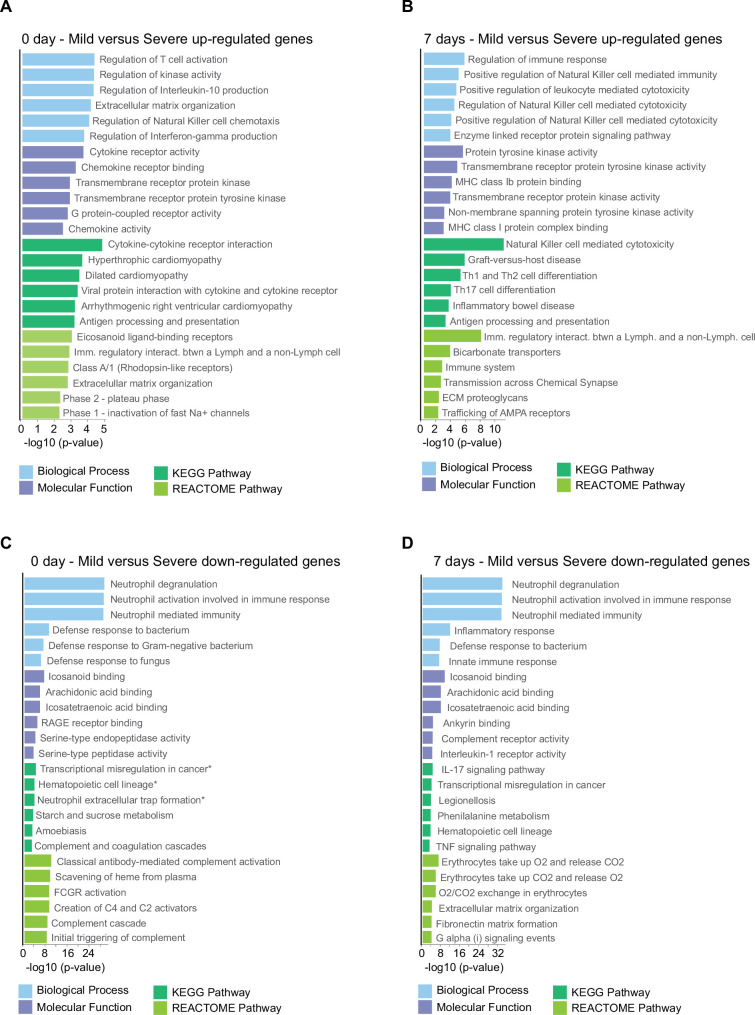
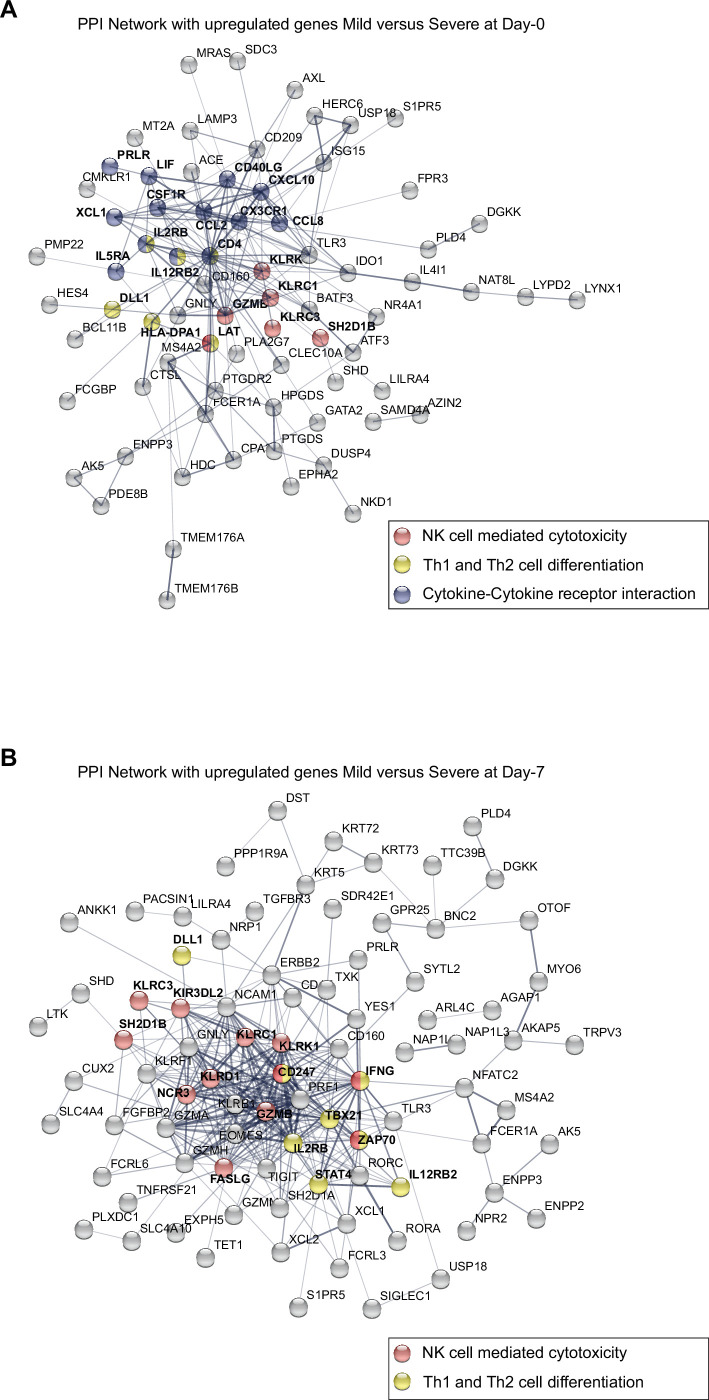
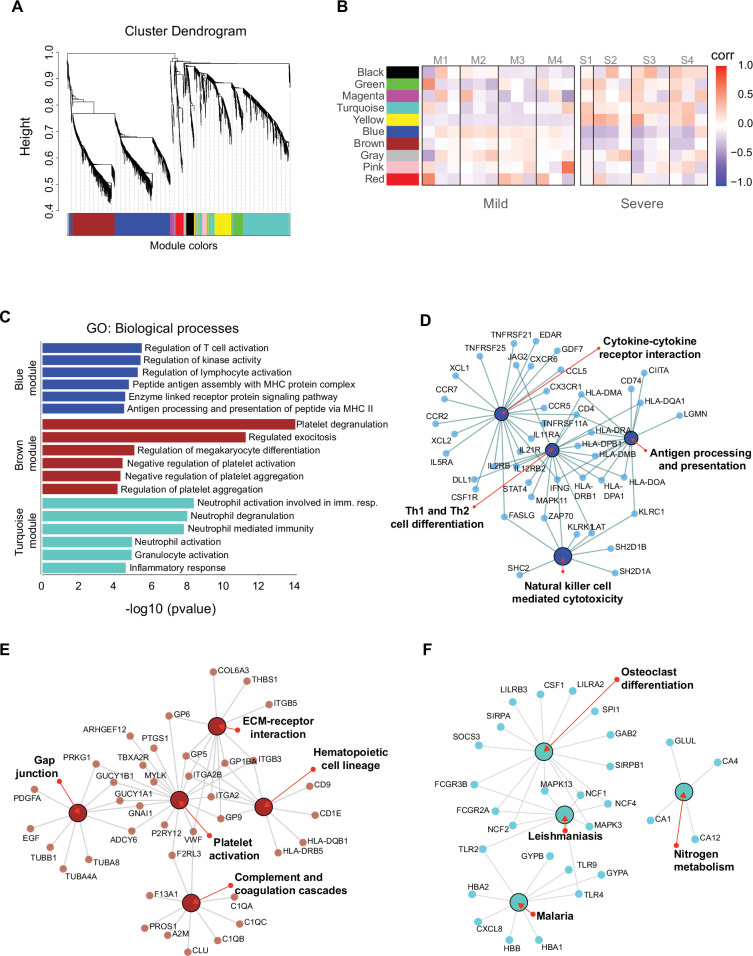
Patients present a wide range of clinical severities in response severe acute respiratory syndrome coronavirus 2 infection, but the underlying molecular and cellular reasons why clinical outcomes vary so greatly within the population remains unknown. Here, we report that negative clinical outcomes in severely ill patients were associated with divergent RNA transcriptome profiles in peripheral immune cells compared with mild cases during the first weeks after disease onset. Protein-protein interaction analysis indicated that early-responding cytotoxic natural killer cells were associated with an effective clearance of the virus and a less severe outcome. This innate immune response was associated with the activation of select cytokine-cytokine receptor pathways and robust Th1/Th2 cell differentiation profiles. In contrast, severely ill patients exhibited a dysregulation between innate and adaptive responses affiliated with divergent Th1/Th2 profiles and negative outcomes. This knowledge forms the basis of clinical triage that may be used to preemptively detect high-risk patients before life-threatening outcomes ensue.
Keywords: COVID-19; NK; PBMC RNA-seq; human; infectious disease; microbiology.
© 2024, Medina et al.
Conflict of interest statement
MM, FF, CQ, FA, RR, MR, SB, YP, MC, IB, CK, RA, JG, MB No competing interests declared
Figures




Update of
- doi: 10.1101/2024.01.02.573936
- doi: 10.7554/eLife.94242.1
- doi: 10.7554/eLife.94242.2
References
-
- Ahern DJ, Ai Z, Ainsworth M, Allan C, Allcock A, Angus B, Ansari MA, Arancibia-Cárcamo CV, Aschenbrenner D, Attar M, Baillie JK, Barnes E, Bashford-Rogers R, Bashyal A, Beer S, Berridge G, Beveridge A, Bibi S, Bicanic T, Blackwell L, Bowness P, Brent A, Brown A, Broxholme J, Buck D, Burnham KL, Byrne H, Camara S, Candido Ferreira I, Charles P, Chen W, Chen Y-L, Chong A, Clutterbuck EA, Coles M, Conlon CP, Cornall R, Cribbs AP, Curion F, Davenport EE, Davidson N, Davis S, Dendrou CA, Dequaire J, Dib L, Docker J, Dold C, Dong T, Downes D, Drakesmith H, Dunachie SJ, Duncan DA, Eijsbouts C, Esnouf R, Espinosa A, Etherington R, Fairfax B, Fairhead R, Fang H, Fassih S, Felle S, Fernandez Mendoza M, Ferreira R, Fischer R, Foord T, Forrow A, Frater J, Fries A, Gallardo Sanchez V, Garner LC, Geeves C, Georgiou D, Godfrey L, Golubchik T, Gomez Vazquez M, Green A, Harper H, Harrington HA, Heilig R, Hester S, Hill J, Hinds C, Hird C, Ho L-P, Hoekzema R, Hollis B, Hughes J, Hutton P, Jackson-Wood MA, Jainarayanan A, James-Bott A, Jansen K, Jeffery K, Jones E, Jostins L, Kerr G, Kim D, Klenerman P, Knight JC, Kumar V, Kumar Sharma P, Kurupati P, Kwok A, Lee A, Linder A, Lockett T, Lonie L, Lopopolo M, Lukoseviciute M, Luo J, Marinou S, Marsden B, Martinez J, Matthews PC, Mazurczyk M, McGowan S, McKechnie S, Mead A, Mentzer AJ, Mi Y, Monaco C, Montadon R, Napolitani G, Nassiri I, Novak A, O’Brien DP, O’Connor D, O’Donnell D, Ogg G, Overend L, Park I, Pavord I, Peng Y, Penkava F, Pereira Pinho M, Perez E, Pollard AJ, Powrie F, Psaila B, Quan TP, Repapi E, Revale S, Silva-Reyes L, Richard J-B, Rich-Griffin C, Ritter T, Rollier CS, Rowland M, Ruehle F, Salio M, Sansom SN, Sanches Peres R, Santos Delgado A, Sauka-Spengler T, Schwessinger R, Scozzafava G, Screaton G, Seigal A, Semple MG, Sergeant M, Simoglou Karali C, Sims D, Skelly D, Slawinski H, Sobrinodiaz A, Sousos N, Stafford L, Stockdale L, Strickland M, Sumray O, Sun B, Taylor C, Taylor S, Taylor A, Thongjuea S, Thraves H, Todd JA, Tomic A, Tong O, Trebes A, Trzupek D, Tucci FA, Turtle L, Udalova I, Uhlig H, van Grinsven E, Vendrell I, Verheul M, Voda A, Wang G, Wang L, Wang D, Watkinson P, Watson R, Weinberger M, Whalley J, Witty L, Wray K, Xue L, Yeung HY, Yin Z, Young RK, Youngs J, Zhang P, Zurke Y-X. A blood atlas of COVID-19 defines hallmarks of disease severity and specificity. Cell. 2022;185:916–938. doi: 10.1016/j.cell.2022.01.012. - DOI - PMC - PubMed
-
- Andrews S. FastQC. GPL v3Github. 2010 https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
- Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann H-H, Zhang Y, Dorgham K, Philippot Q, Rosain J, Béziat V, Manry J, Shaw E, Haljasmägi L, Peterson P, Lorenzo L, Bizien L, Trouillet-Assant S, Dobbs K, de Jesus AA, Belot A, Kallaste A, Catherinot E, Tandjaoui-Lambiotte Y, Le Pen J, Kerner G, Bigio B, Seeleuthner Y, Yang R, Bolze A, Spaan AN, Delmonte OM, Abers MS, Aiuti A, Casari G, Lampasona V, Piemonti L, Ciceri F, Bilguvar K, Lifton RP, Vasse M, Smadja DM, Migaud M, Hadjadj J, Terrier B, Duffy D, Quintana-Murci L, van de Beek D, Roussel L, Vinh DC, Tangye SG, Haerynck F, Dalmau D, Martinez-Picado J, Brodin P, Nussenzweig MC, Boisson-Dupuis S, Rodríguez-Gallego C, Vogt G, Mogensen TH, Oler AJ, Gu J, Burbelo PD, Cohen JI, Biondi A, Bettini LR, D’Angio M, Bonfanti P, Rossignol P, Mayaux J, Rieux-Laucat F, Husebye ES, Fusco F, Ursini MV, Imberti L, Sottini A, Paghera S, Quiros-Roldan E, Rossi C, Castagnoli R, Montagna D, Licari A, Marseglia GL, Duval X, Ghosn J, HGID Lab. NIAID-USUHS Immune Response to COVID Group. COVID Clinicians. COVID-STORM Clinicians. Imagine COVID Group. French COVID Cohort Study Group. Milieu Intérieur Consortium. CoV-Contact Cohort. Amsterdam UMC Covid-19 Biobank. COVID Human Genetic Effort. Tsang JS, Goldbach-Mansky R, Kisand K, Lionakis MS, Puel A, Zhang S-Y, Holland SM, Gorochov G, Jouanguy E, Rice CM, Cobat A, Notarangelo LD, Abel L, Su HC, Casanova J-L. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:eabd4585. doi: 10.1126/science.abd4585. - DOI - PMC - PubMed
-
- Bernal E, Gimeno L, Alcaraz MJ, Quadeer AA, Moreno M, Martínez-Sánchez MV, Campillo JA, Gomez JM, Pelaez A, García E, Herranz M, Hernández-Olivo M, Martínez-Alfaro E, Alcaraz A, Muñoz Á, Cano A, McKay MR, Muro M, Minguela A. Activating killer-cell immunoglobulin-like receptors are associated with the severity of coronavirus disease 2019. The Journal of Infectious Diseases. 2021;224:229–240. doi: 10.1093/infdis/jiab228. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
