

Homozygous variants in WDR83OS lead to a neurodevelopmental disorder with hypercholanemia
- PMID: 39471804
- PMCID: PMC11568760
- DOI: 10.1016/j.ajhg.2024.10.002
Homozygous variants in WDR83OS lead to a neurodevelopmental disorder with hypercholanemia
Abstract
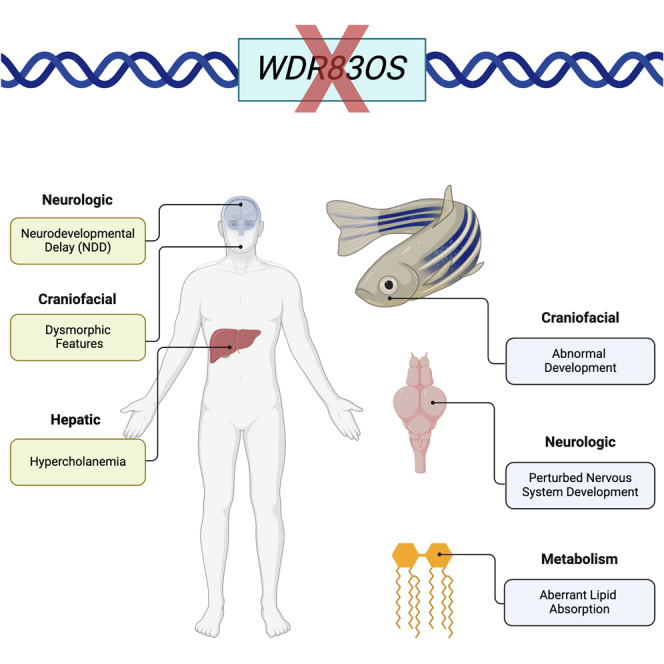
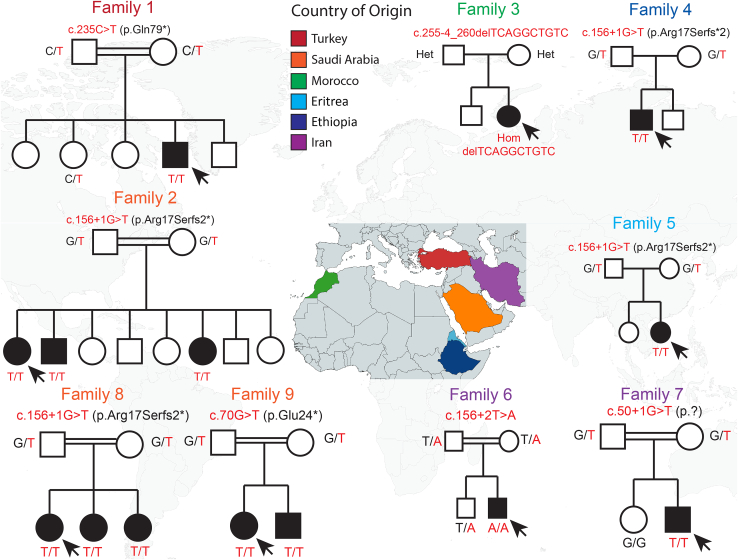
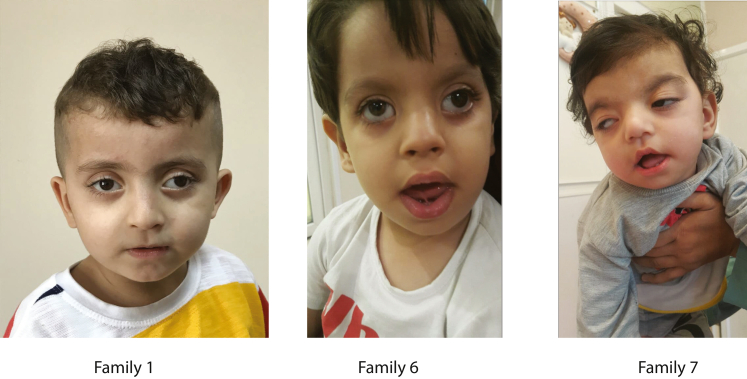
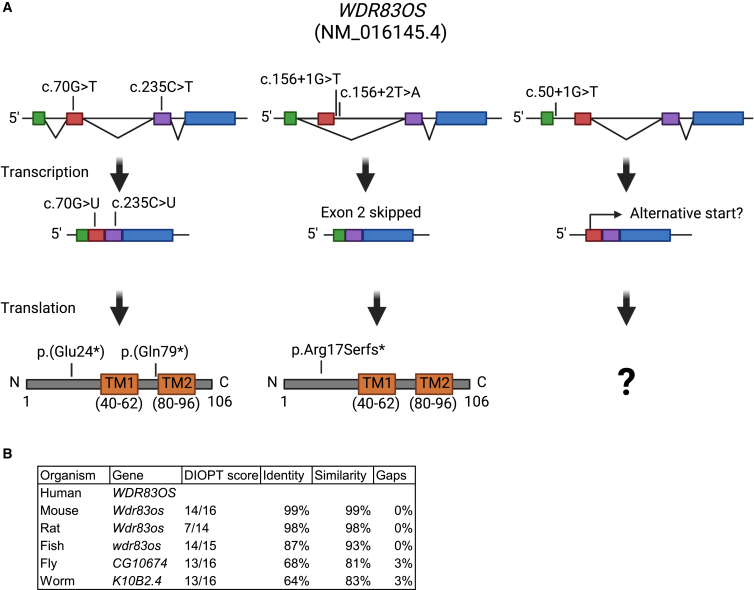
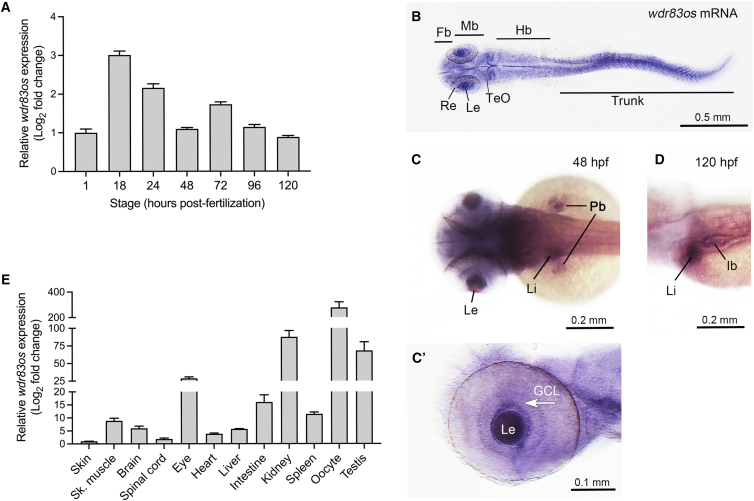
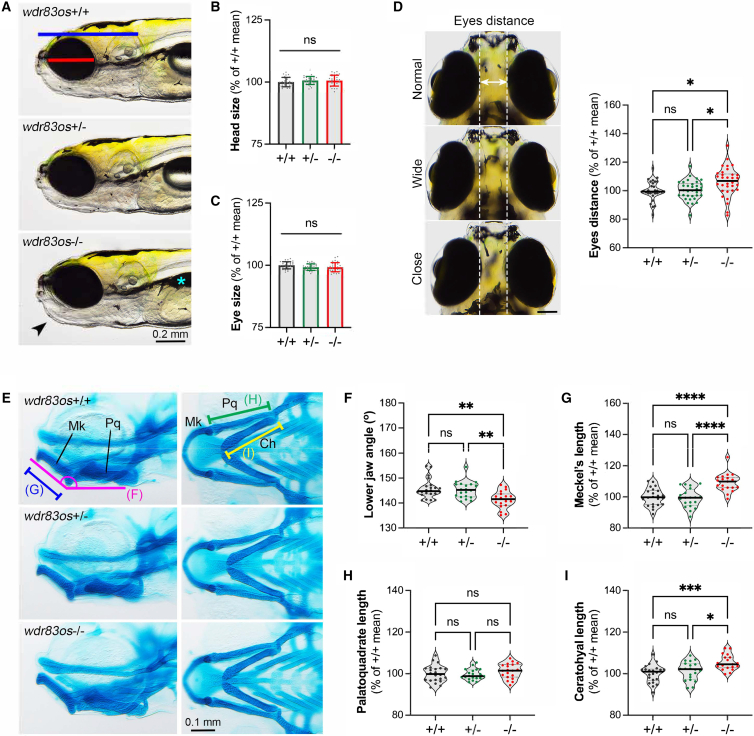
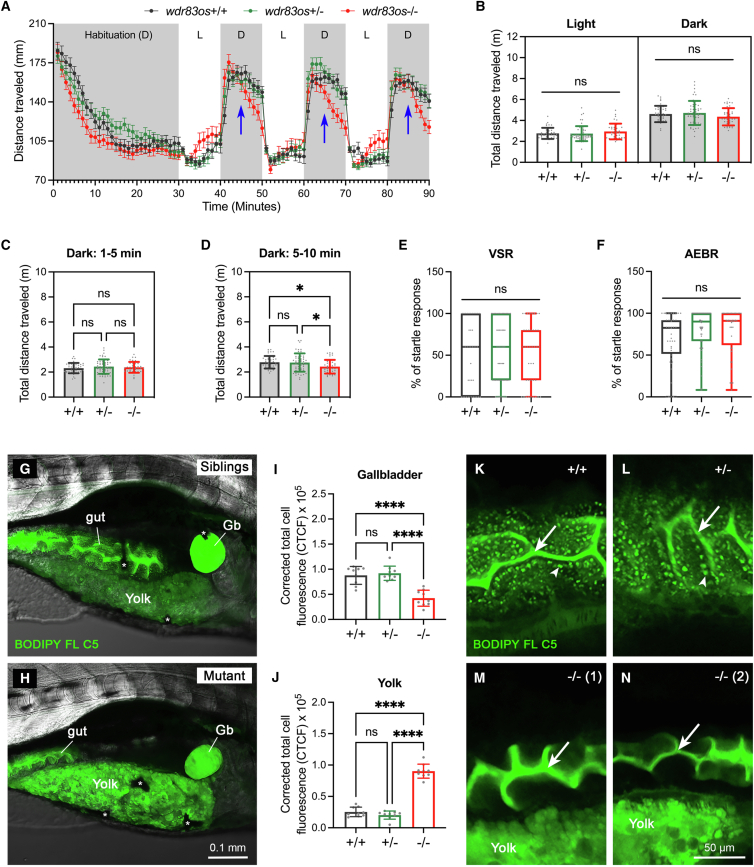
WD repeat domain 83 opposite strand (WDR83OS) encodes the 106-aa (amino acid) protein Asterix, which heterodimerizes with CCDC47 to form the PAT (protein associated with ER translocon) complex. This complex functions as a chaperone for large proteins containing transmembrane domains to ensure proper folding. Until recently, little was known about the role of WDR83OS or CCDC47 in human disease traits. However, biallelic variants in CCDC47 were identified in four unrelated families with trichohepatoneurodevelopmental syndrome, characterized by a neurodevelopmental disorder (NDD) with liver dysfunction. Three affected siblings in an additional family share a homozygous truncating WDR83OS variant and a phenotype of NDD, dysmorphic features, and liver dysfunction. Using family-based rare variant analyses of exome sequencing (ES) data and case matching through GeneMatcher, we describe the clinical phenotypes of 11 additional individuals in eight unrelated families (nine unrelated families, 14 individuals in total) with biallelic putative truncating variants in WDR83OS. Consistent clinical features include NDD (14/14), facial dysmorphism (13/14), intractable itching (9/14), and elevated bile acids (5/6). Whereas bile acids were significantly elevated in 5/6 of individuals tested, bilirubin was normal and liver enzymes were normal to mildly elevated in all 14 individuals. In three of six individuals for whom longitudinal data were available, we observed a progressive reduction in relative head circumference. A zebrafish model lacking Wdr83os function further supports its role in the nervous system, craniofacial development, and lipid absorption. Taken together, our data support a disease-gene association between biallelic loss-of-function of WDR83OS and a neurological disease trait with hypercholanemia.
Keywords: ASTERIX; CCDC47; ER translocation; PAT complex; WDR83OS; developmental delay; hypercholanemia; intellectual disability; pruritus.
Copyright © 2024 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The Department of Molecular & Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics Laboratories. J.R.L. serves on the Scientific Advisory Board of Baylor Genetics. J.R.L. has stock ownership in 23andMe, is a paid consultant for Genome International, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting.
Figures
References
-
- Alfadhel M., Umair M., Almuzzaini B., Asiri A., Al Tuwaijri A., Alhamoudi K., Alyafee Y., Al-Owain M. Identification of the TTC26 Splice Variant in a Novel Complex Ciliopathy Syndrome with Biliary, Renal, Neurological, and Skeletal Manifestations. Mol. Syndromol. 2021;12:133–140. doi: 10.1159/000513829. - DOI - PMC - PubMed
-
- Shaheen R., Alsahli S., Ewida N., Alzahrani F., Shamseldin H.E., Patel N., Al Qahtani A., Alhebbi H., Alhashem A., Al-Sheddi T., et al. Biallelic Mutations in Tetratricopeptide Repeat Domain 26 (Intraflagellar Transport 56) Cause Severe Biliary Ciliopathy in Humans. Hepatology. 2020;71:2067–2079. doi: 10.1002/hep.30982. - DOI - PubMed
-
- David O., Eskin-Schwartz M., Ling G., Dolgin V., Kristal E., Benkowitz E., Osyntsov L., Gradstein L., Birk O.S., Loewenthal N., Yerushalmi B. Pituitary stalk interruption syndrome broadens the clinical spectrum of the TTC26 ciliopathy. Clin. Genet. 2020;98:303–307. doi: 10.1111/cge.13805. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
