

A national long-read sequencing study on chromosomal rearrangements uncovers hidden complexities
- PMID: 39472022
- PMCID: PMC11610602
- DOI: 10.1101/gr.279510.124
A national long-read sequencing study on chromosomal rearrangements uncovers hidden complexities
Abstract
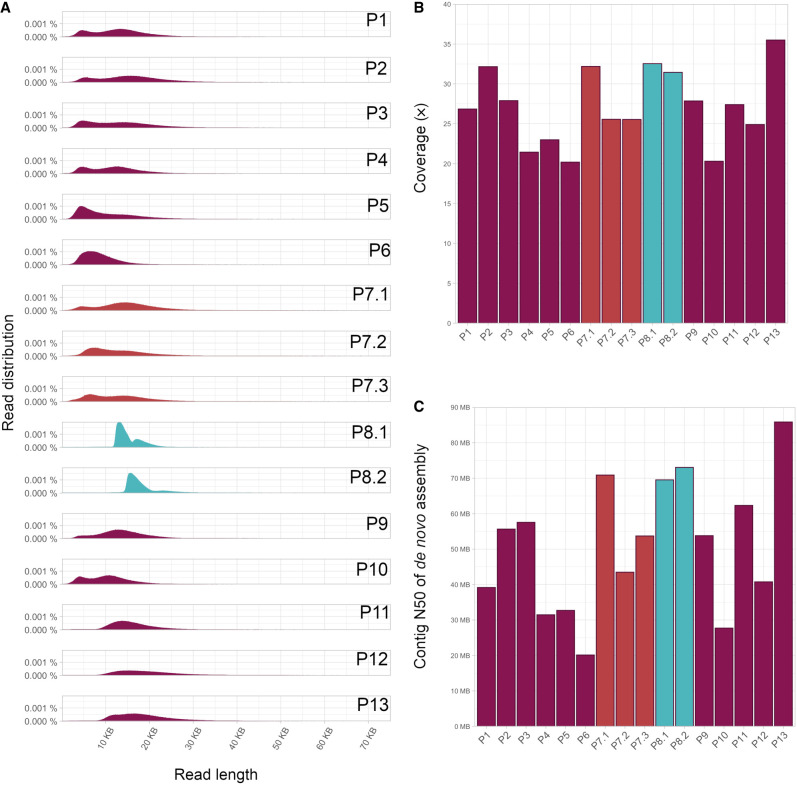
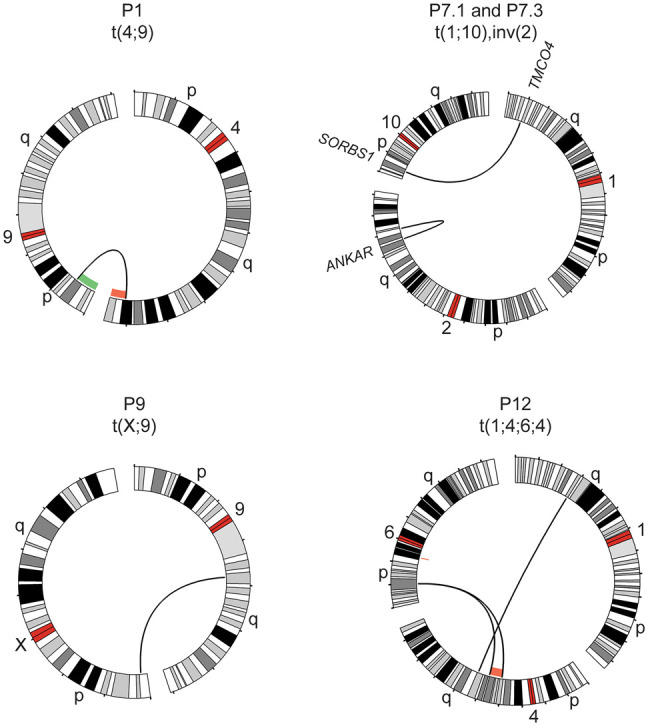
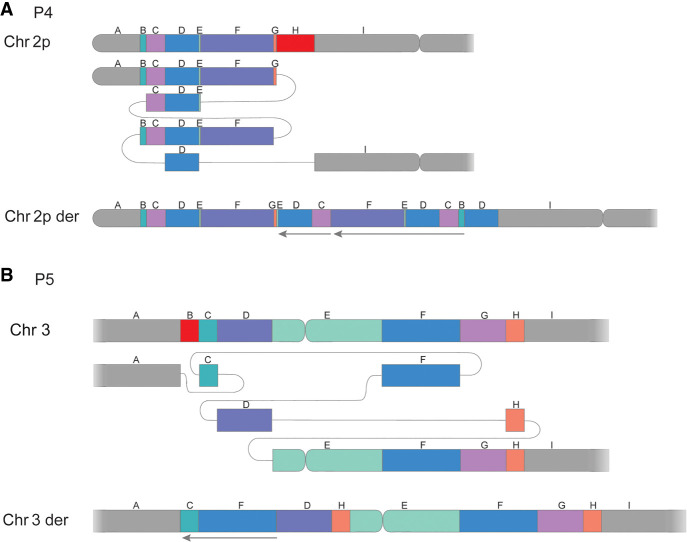
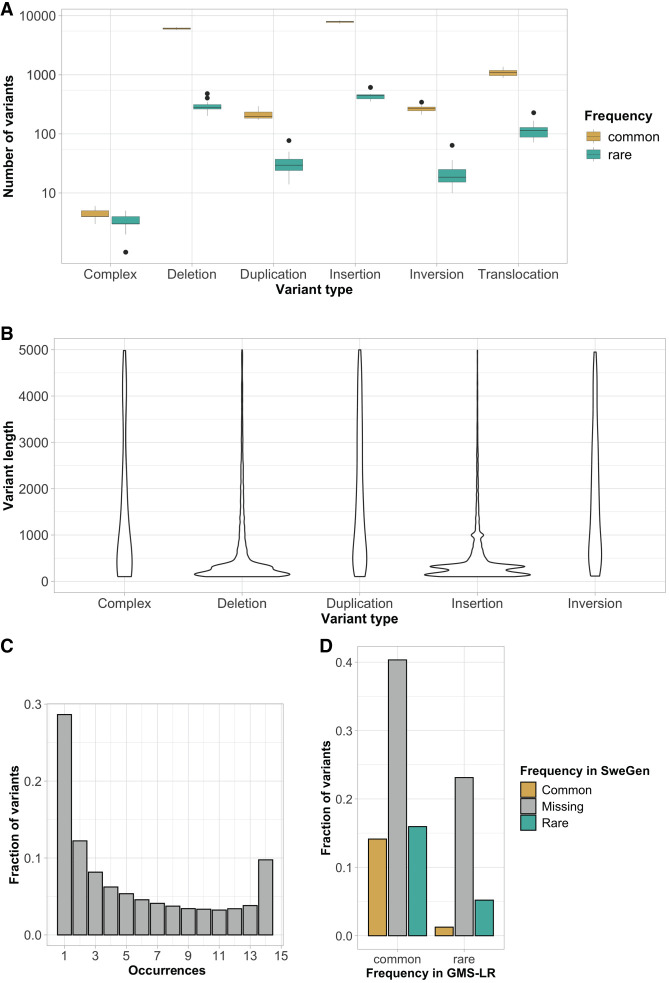
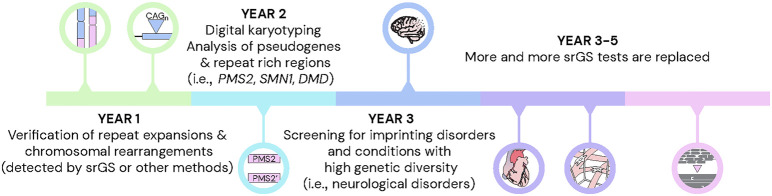
Clinical genetic laboratories often require a comprehensive analysis of chromosomal rearrangements/structural variants (SVs), from large events like translocations and inversions to supernumerary ring/marker chromosomes and small deletions or duplications. Understanding the complexity of these events and their clinical consequences requires pinpointing breakpoint junctions and resolving the derivative chromosome structure. This task often surpasses the capabilities of short-read sequencing technologies. In contrast, long-read sequencing techniques present a compelling alternative for clinical diagnostics. Here, Genomic Medicine Sweden-Rare Diseases has explored the utility of HiFi Revio long-read genome sequencing (lrGS) for digital karyotyping of SVs nationwide. The 16 samples from 13 families were collected from all Swedish healthcare regions. Prior investigations had identified 16 SVs, ranging from simple to complex rearrangements, including inversions, translocations, and copy number variants. We have established a national pipeline and a shared variant database for variant calling and filtering. Using lrGS, 14 of the 16 known SVs are detected. Of these, 13 are mapped at nucleotide resolution, and one complex rearrangement is only visible by read depth. Two Chromosome 21 rearrangements, one mosaic, remain undetected. Average read lengths are 8.3-18.8 kb with coverage exceeding 20× for all samples. De novo assembly results in a limited number of phased contigs per individual (N50 6-86 Mb), enabling direct characterization of the chromosomal rearrangements. In a national pilot study, we demonstrate the utility of HiFi Revio lrGS for analyzing chromosomal rearrangements. Based on our results, we propose a 5-year plan to expand lrGS use for rare disease diagnostics in Sweden.
© 2024 Eisfeldt et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous