

From emergence to endemicity of highly pathogenic H5 avian influenza viruses in Taiwan
- PMID: 39472594
- PMCID: PMC11522503
- DOI: 10.1038/s41467-024-53816-y
From emergence to endemicity of highly pathogenic H5 avian influenza viruses in Taiwan
Abstract
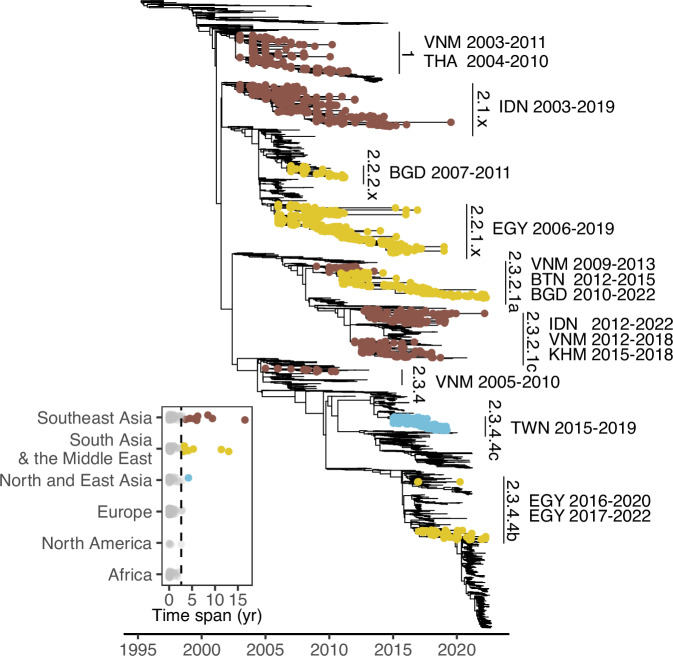
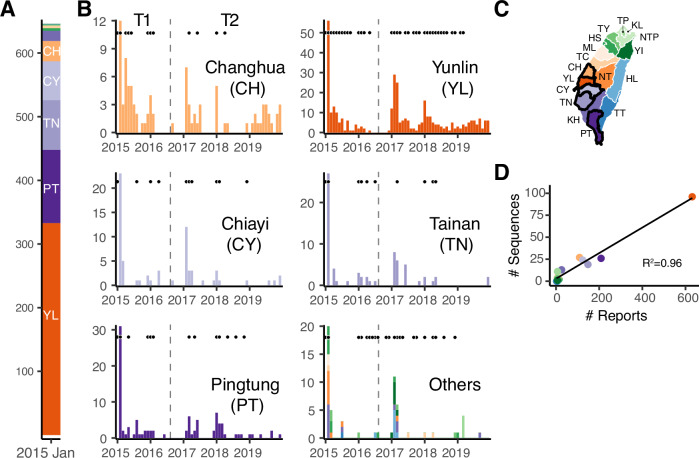
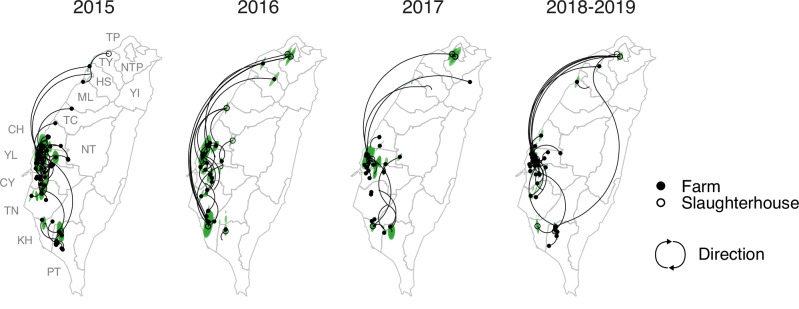
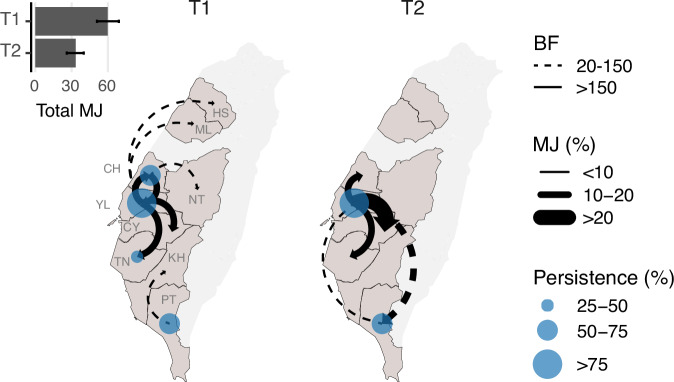
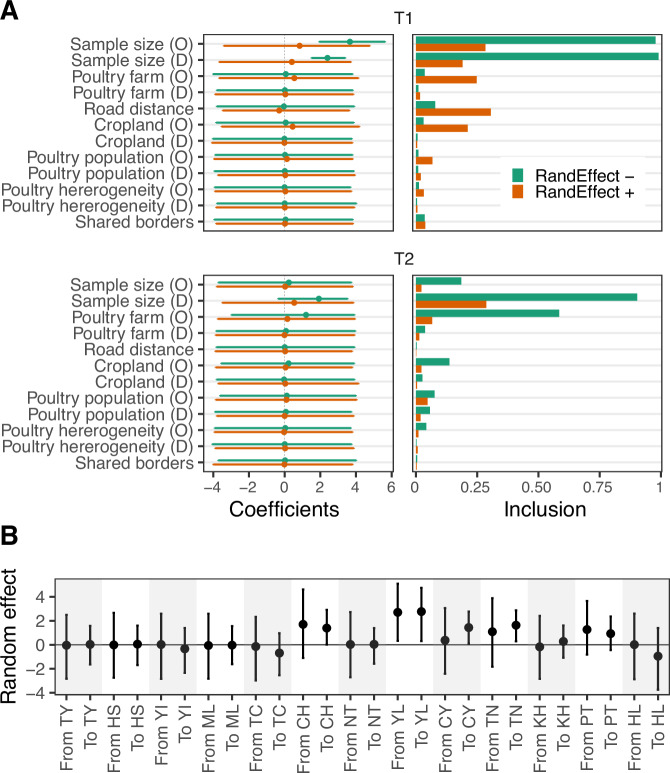
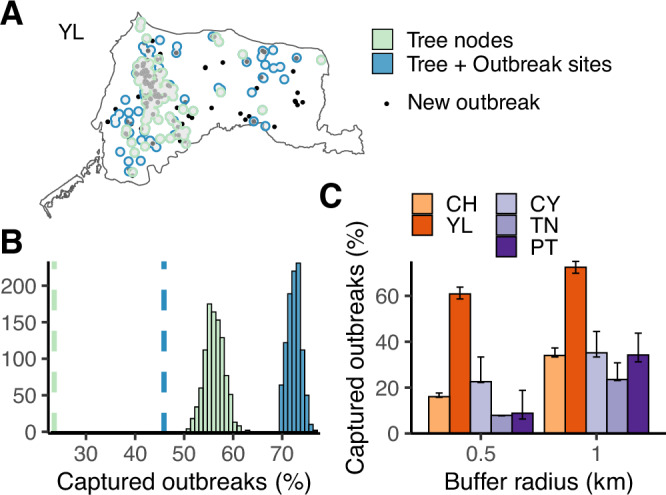
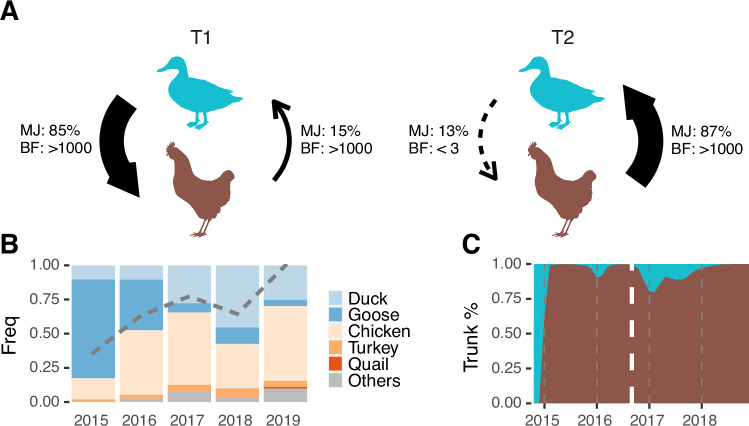
A/goose/Guangdong/1/96-like (GsGd) highly pathogenic avian influenza (HPAI) H5 viruses cause severe outbreaks in poultry when introduced. Since emergence in 1996, control measures in most countries have suppressed local GsGd transmission following introductions, making persistent transmission in domestic birds rare. However, geographical expansion of clade 2.3.4.4 sublineages has raised concern about establishment of endemic circulation, while mechanistic drivers leading to endemicity remain unknown. We reconstructed the evolutionary history of GsGd sublineage, clade 2.3.4.4c, in Taiwan using a time-heterogeneous rate phylogeographic model. During Taiwan's initial epidemic wave (January 2015 - August 2016), we inferred that localised outbreaks had multiple origins from rapid spread between counties/cities nationwide. Subsequently, outbreaks predominantly originated from a single county, Yunlin, where persistent transmission harbours the trunk viruses of the sublineage. Endemic hotspots determined by phylogeographic reconstruction largely predicted the locations of re-emerging outbreaks in Yunlin. The transition to endemicity involved a shift to chicken-dominant circulation, following the initial bidirectional spread between chicken and domestic waterfowl. Our results suggest that following their emergence in Taiwan, source-sink dynamics from a single county have maintained GsGd endemicity up until 2023, pointing to where control efforts should be targeted to eliminate the disease.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Smith, G. J. D., Donis, R. O. & World Health Organization/World. Organisation for Animal Health/Food and Agriculture Organization (WHO/OIE/FAO) H5 Evolution Working Group. Nomenclature updates resulting from the evolution of avian influenza A(H5) virus clades 2.1.3.2a, 2.2.1, and 2.3.4 during 2013–2014. Influenza Other Respir. Viruses9, 271–276 (2015). - PMC - PubMed
-
- FAO. Approaches to controlling, preventing and eliminating H5N1 Highly Pathogenic Avian Influenza in endemic countries. Animal Production and Health Paper. No. 171, Rome (2011).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
