

Promoting readthrough of nonsense mutations in CF mouse model: Biodistribution and efficacy of NV848 in rescuing CFTR protein expression
- PMID: 39473179
- PMCID: PMC11638873
- DOI: 10.1016/j.ymthe.2024.10.028
Promoting readthrough of nonsense mutations in CF mouse model: Biodistribution and efficacy of NV848 in rescuing CFTR protein expression
Abstract
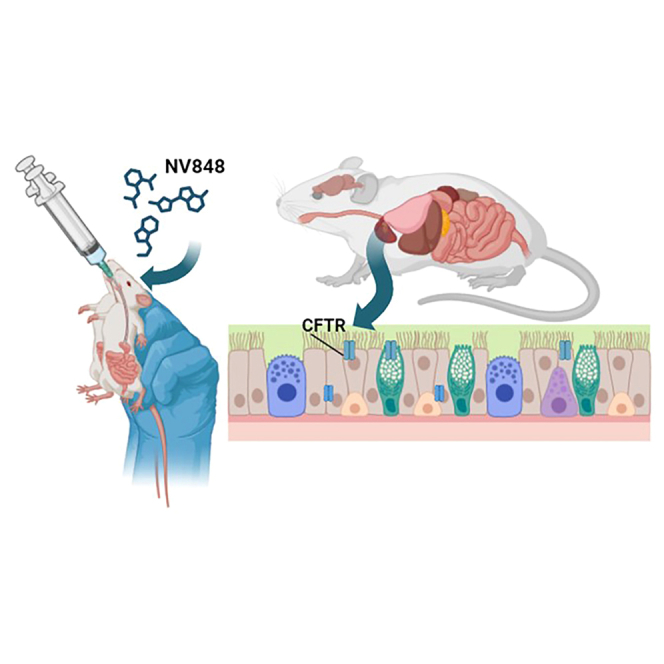
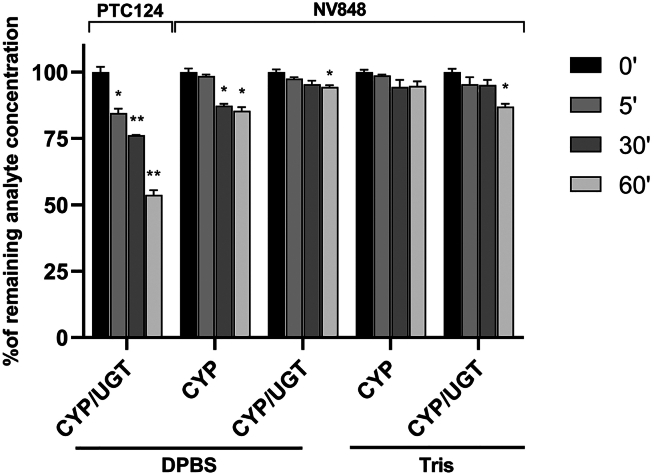
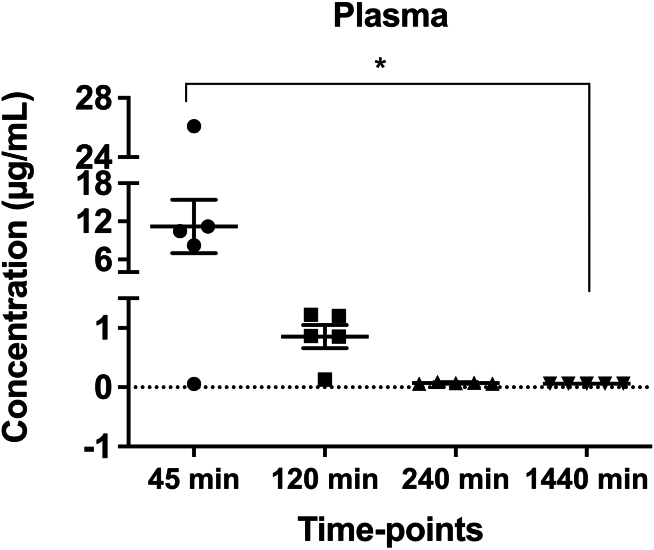
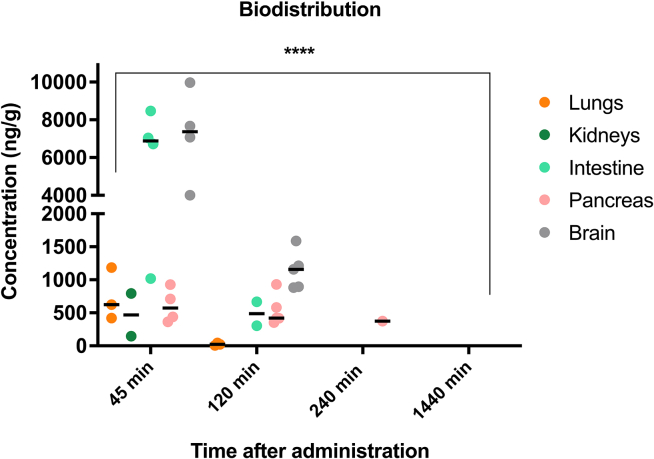
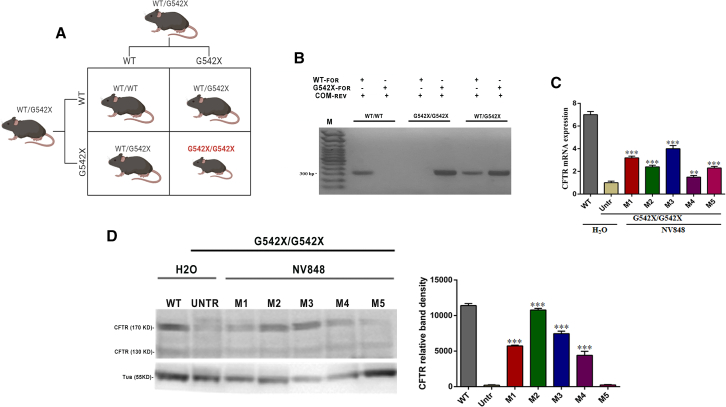
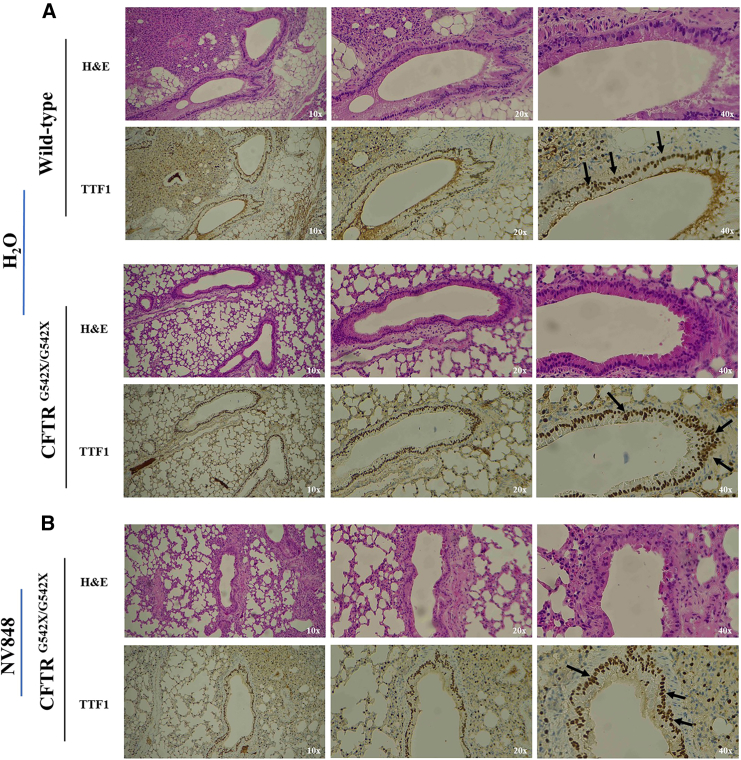
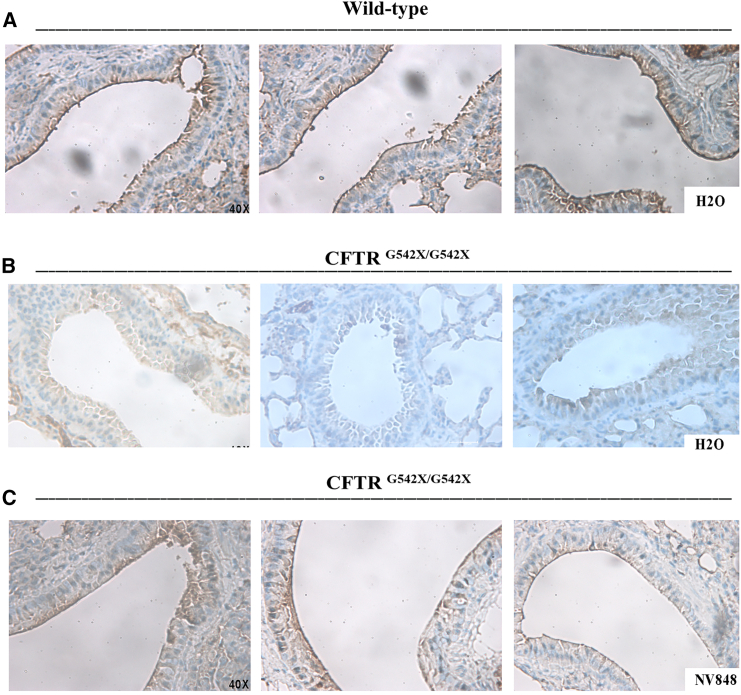
Nonsense mutations, often resulting from single-nucleotide substitutions, produce mRNA harboring a premature termination codon (PTC), which causes the premature termination of protein synthesis. This produces truncated and non-functional proteins, which cause different genetic diseases, including cystic fibrosis (CF). This work aims to investigate the ability of NV848 (N-(5-methyl-1,2,4-oxadiazol-3-yl)acetamide), a translational readthrough-inducing drug (TRID), to rescue CF transmembrane conductance regulator (CFTR) protein expression in a murine model characterized by the G542X nonsense mutation in the CFTR gene. In vitro experiments assessed the drug's stability in human hepatic metabolism, and in vivo investigations on wild-type mice allowed us to clarify the distribution of the drug to the target organs. Moreover, its efficacy in recovering the CFTR protein after chronic treatment was assessed in G542X homozygous mice. Our results provide valuable insights into the biodistribution and therapeutic attributes of NV848, representing a promising therapeutic tool for enhanced clinical outcomes in individuals affected by CF with nonsense mutations.
Keywords: Mendelian diseases; cystic fibrosis; nonsense; oxadiazole; stop codons.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests I.P. is a scientific advisor of the CCM Bioscience group. I.P., L.L., A.P., M.T., and R.M. have patent licenses to WO2019101709.
Figures
References
-
- Clarke L.A., Awatade N.T., Felício V.M., Silva I.A., Calucho M., Pereira L., Azevedo P., Cavaco J., Barreto C., Bertuzzo C., et al. The effect of premature termination codon mutations on CFTR mRNA abundance in human nasal epithelium and intestinal organoids: a basis for read-through therapies in cystic fibrosis. Hum. Mutat. 2019;40:326–334. doi: 10.1002/humu.23692. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
