

Understanding Voriconazole Metabolism: A Middle-Out Physiologically-Based Pharmacokinetic Modelling Framework Integrating In Vitro and Clinical Insights
- PMID: 39476315
- PMCID: PMC11573852
- DOI: 10.1007/s40262-024-01434-8
Understanding Voriconazole Metabolism: A Middle-Out Physiologically-Based Pharmacokinetic Modelling Framework Integrating In Vitro and Clinical Insights
Abstract
Background and objective: Voriconazole (VRC), a broad-spectrum antifungal drug, exhibits nonlinear pharmacokinetics (PK) due to saturable metabolic processes, autoinhibition and metabolite-mediated inhibition on their own formation. VRC PK is also characterised by high inter- and intraindividual variability, primarily associated with cytochrome P450 (CYP) 2C19 genetic polymorphism. Additionally, recent in vitro findings indicate that VRC main metabolites, voriconazole N-oxide (NO) and hydroxyvoriconazole (OHVRC), inhibit CYP enzymes responsible for VRC metabolism, adding to its PK variability. This variability poses a significant risk of therapeutic failure or adverse events, which are major challenges in VRC therapy. Understanding the underlying processes and sources of these variabilities is essential for safe and effective therapy. This work aimed to develop a whole-body physiologically-based pharmacokinetic (PBPK) modelling framework that elucidates the complex metabolism of VRC and the impact of its metabolites, NO and OHVRC, on the PK of the parent, leveraging both in vitro and in vivo clinical data in a middle-out approach.
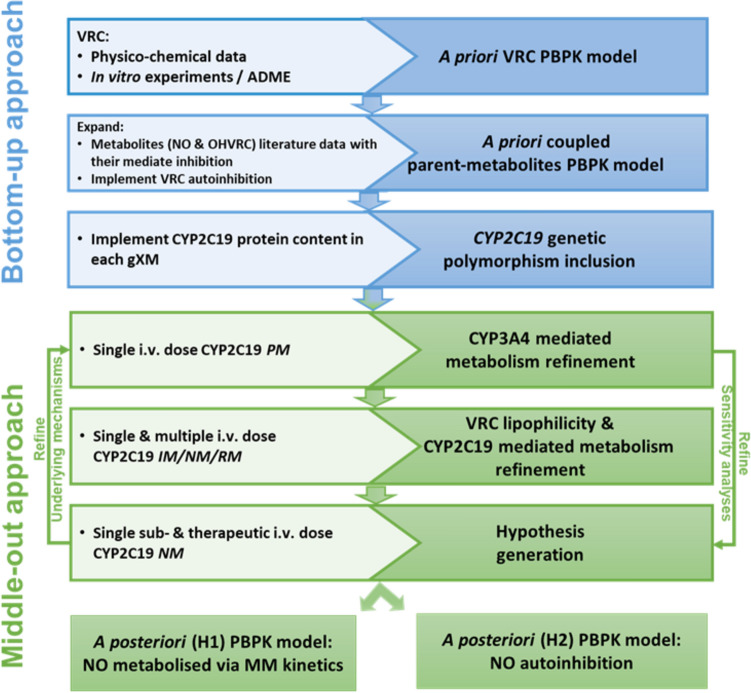
Methods: A coupled parent-metabolite PBPK model for VRC, NO and OHVRC was developed in a stepwise manner using PK-Sim® and MoBi®. Based on available in vitro data, NO formation was assumed to be mediated by CYP2C19, CYP3A4, and CYP2C9, while OHVRC formation was attributed solely to CYP3A4. Both metabolites were assumed to be excreted via renal clearance, with hepatic elimination also considered for NO. Inhibition functions were implemented to describe the complex interaction network of VRC autoinhibition and metabolite-mediated inhibition on each CYP enzyme.
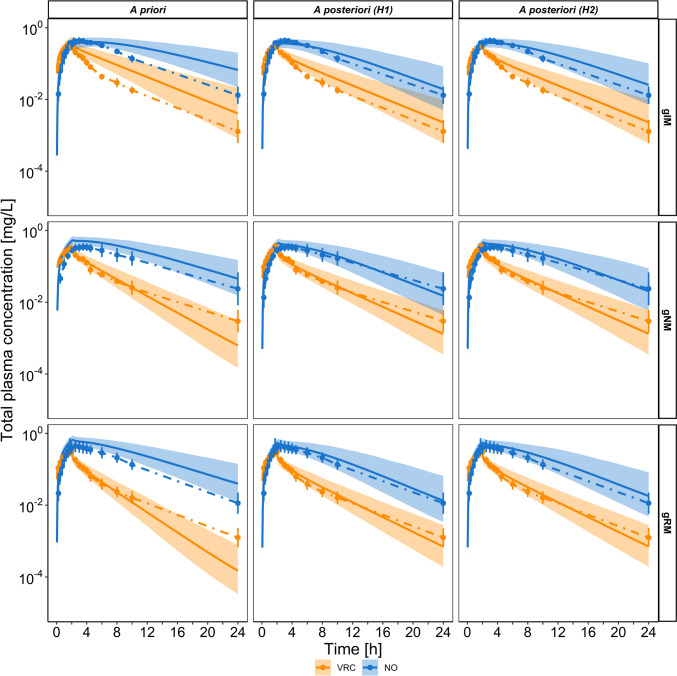
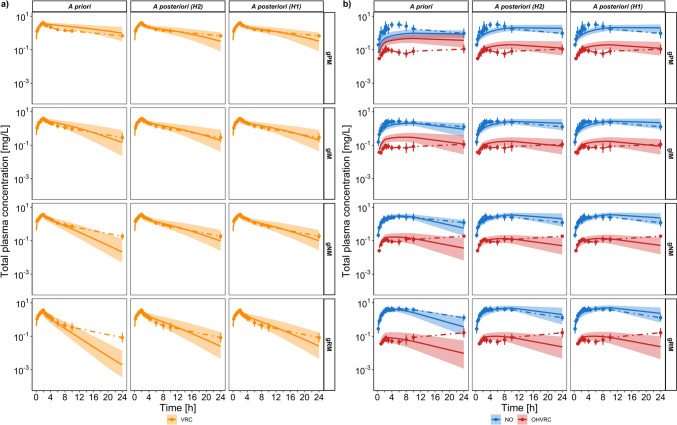
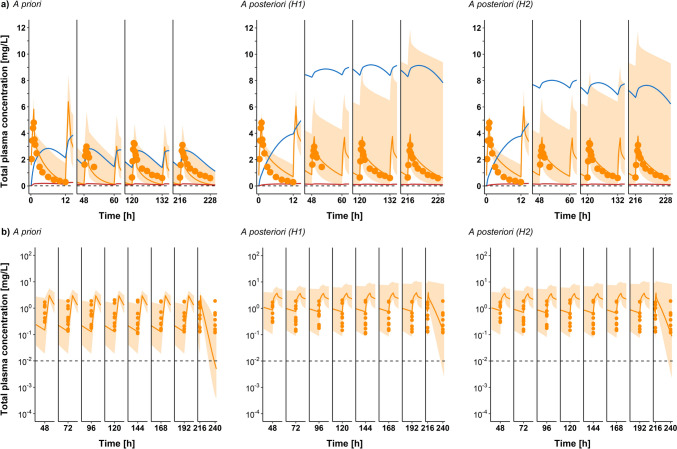
Results: Using a combined bottom-up and middle-out approach, incorporating data from multiple clinical studies and existing literature, the model accurately predicted plasma concentration-time profiles across various intravenous dosing regimens in healthy adults, of different CYP2C19 genotype-predicted phenotypes. All (100%) of the predicted area under the concentration-time curve (AUC) and 94% of maximum concentration (Cmax) values of VRC met the 1.25-fold acceptance criterion, with overall absolute average fold errors of 1.12 and 1.14, respectively. Furthermore, all predicted AUC and Cmax values of NO and OHVRC met the twofold acceptance criterion.
Conclusion: This comprehensive parent-metabolite PBPK model of VRC quantitatively elucidated the complex metabolism of the drug and emphasised the substantial impact of the primary metabolites on VRC PK. The comprehensive approach combining bottom-up and middle-out modelling, thereby accounting for VRC autoinhibition, metabolite-mediated inhibition, and the impact of CYP2C19 genetic polymorphisms, enhances our understanding of VRC PK. Moreover, the model can be pivotal in designing further in vitro experiments, ultimately allowing for extrapolation to paediatric populations, enhance treatment individualisation and improve clinical outcomes.
© 2024. The Author(s).
Conflict of interest statement
Figures





Comment in
-
Comment on "Understanding Voriconazole Metabolism: A Middle-Out Physiologically-Based Pharmacokinetic Modelling Framework Integrating In Vitro and Clinical Insights".Clin Pharmacokinet. 2025 Sep;64(9):1425-1427. doi: 10.1007/s40262-025-01554-9. Epub 2025 Aug 1. Clin Pharmacokinet. 2025. PMID: 40750726 No abstract available.
References
-
- Perlin DS, Rautemaa-Richardson R, Alastruey-Izquierdo A. The global problem of antifungal resistance: prevalence, mechanisms, and management. Lancet Infect Dis. 2017;17:e383–92. - PubMed
-
- Pfizer. VFEND summary of product characteristics (SmPC). Pfizer; 2020.
-
- Pearson MM, Rogers PD, Cleary JD, Chapman SW. Voriconazole: a new triazole antifungal agent. Ann Pharmacother. 2003;37:420–32. 10.1345/aph.1C261. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
