

Pharmacological restoration of GTP hydrolysis by mutant RAS
- PMID: 39476862
- PMCID: PMC11666464
- DOI: 10.1038/s41586-024-08283-2
Pharmacological restoration of GTP hydrolysis by mutant RAS
Abstract
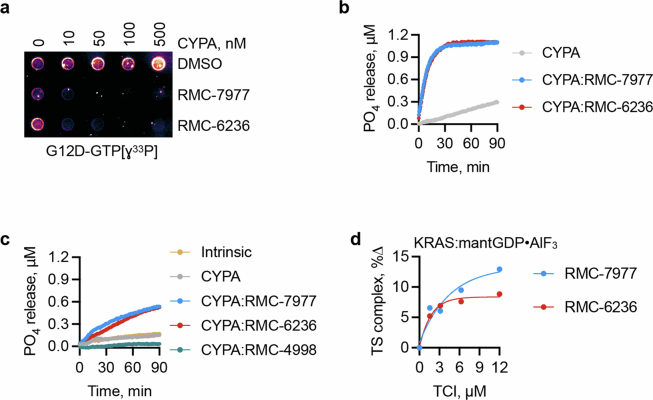
Approximately 3.4 million patients worldwide are diagnosed each year with cancers that have pathogenic mutations in one of three RAS proto-oncogenes (KRAS, NRAS and HRAS)1,2. These mutations impair the GTPase activity of RAS, leading to activation of downstream signalling and proliferation3-6. Long-standing efforts to restore the hydrolase activity of RAS mutants have been unsuccessful, extinguishing any consideration towards a viable therapeutic strategy7. Here we show that tri-complex inhibitors-that is, molecular glues with the ability to recruit cyclophilin A (CYPA) to the active state of RAS-have a dual mechanism of action: not only do they prevent activated RAS from binding to its effectors, but they also stimulate GTP hydrolysis. Drug-bound CYPA complexes modulate residues in the switch II motif of RAS to coordinate the nucleophilic attack on the γ-phosphate of GTP in a mutation-specific manner. RAS mutants that were most sensitive to stimulation of GTPase activity were more susceptible to treatment than mutants in which the hydrolysis could not be enhanced, suggesting that pharmacological stimulation of hydrolysis potentiates the therapeutic effects of tri-complex inhibitors for specific RAS mutants. This study lays the foundation for developing a class of therapeutics that inhibit cancer growth by stimulating mutant GTPase activity.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: P.L. is listed as an inventor on patents filed by MSKCC regarding treatment of KRAS- or BRAF-mutant cancers; reports grants to his institution from Revolution Medicines, Amgen, Mirati and Boehringer Ingelheim; and reports consulting fees or honoraria from Black Diamond Therapeutics, AmMax, OrbiMed, PAQ-Tx, Repare Therapeutics, Boehringer Ingelheim, Menarini Group and Revolution Medicines, as well as membership on the scientific advisory board of Frontier Medicines, Ikena, Biotheryx and PAQ-Tx (consulting fees and equity in each). The other authors declare no competing interests.
Figures













References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
