

Understanding the heterogeneous performance of variant effect predictors across human protein-coding genes
- PMID: 39478110
- PMCID: PMC11526010
- DOI: 10.1038/s41598-024-76202-6
Understanding the heterogeneous performance of variant effect predictors across human protein-coding genes
Abstract
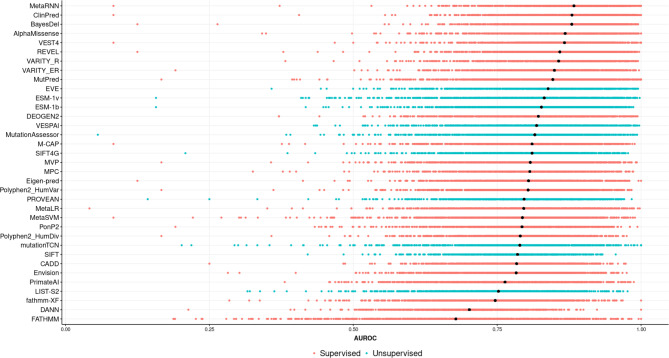
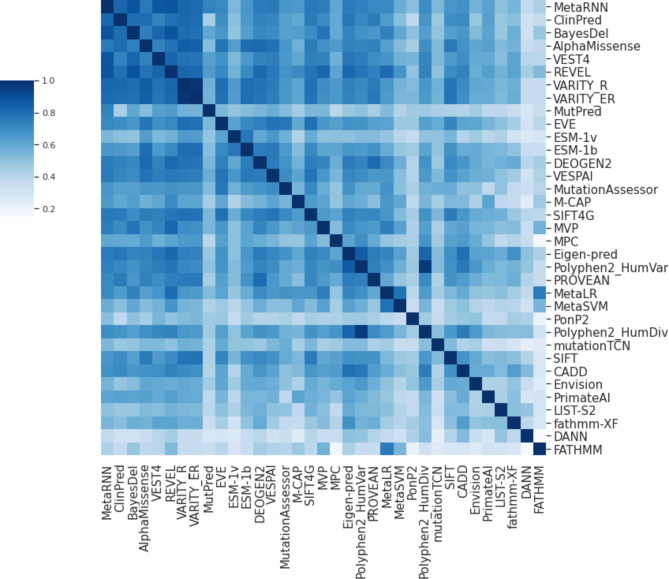
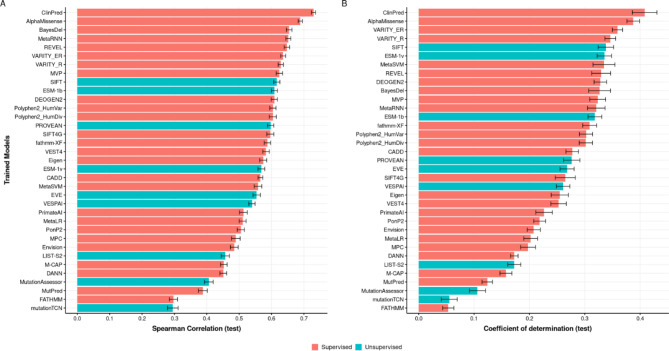
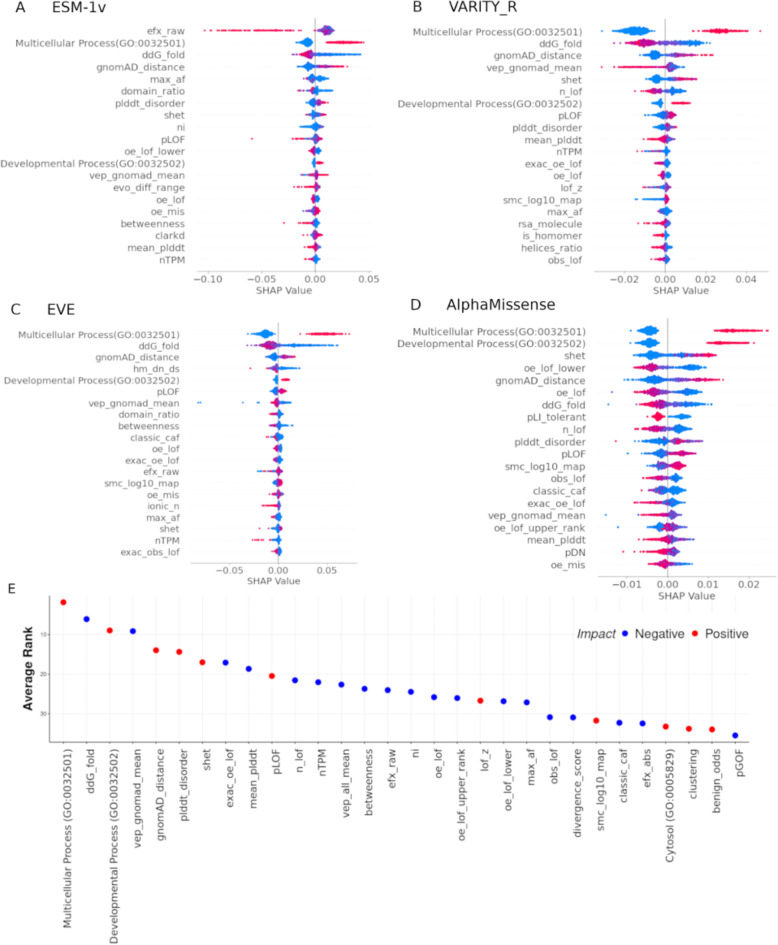
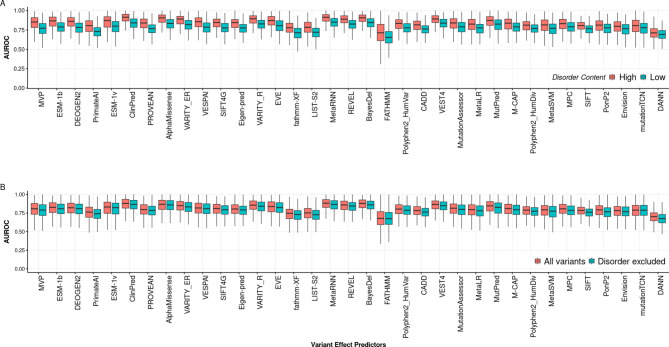
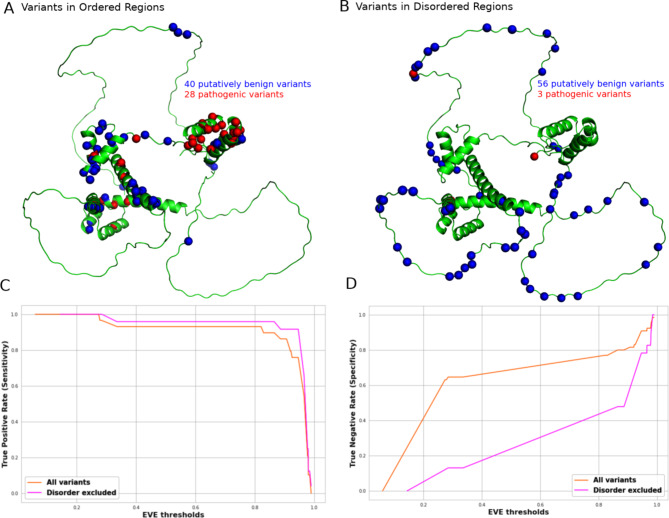
Variant effect predictors (VEPs) are computational tools developed to assess the impacts of genetic mutations, often in terms of likely pathogenicity, employing diverse algorithms and training data. Here, we investigate the performance of 35 VEPs in the discrimination between pathogenic and putatively benign missense variants across 963 human protein-coding genes. We observe considerable gene-level heterogeneity as measured by the widely used area under the receiver operating characteristic curve (AUROC) metric. To investigate the origins of this heterogeneity and the extent to which gene-level VEP performance is predictable, for each VEP, we train random forest models to predict the gene-level AUROC. We find that performance as measured by AUROC is related to factors such as gene function, protein structure, and evolutionary conservation. Notably, intrinsic disorder in proteins emerged as a significant factor influencing apparent VEP performance, often leading to inflated AUROC values due to their enrichment in weakly conserved putatively benign variants. Our results suggest that gene-level features may be useful for identifying genes where VEP predictions are likely to be more or less reliable. However, our work also shows that AUROC, despite being independent of class balance, still has crucial limitations when used for comparing VEP performance across different genes.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Mardis, E. R. Next-generation sequencing platforms. Annu. Rev. Anal. Chem.610.1146/annurev-anchem-062012-092628 (2013). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
