

Differential chromatin accessibility and transcriptional dynamics define breast cancer subtypes and their lineages
- PMID: 39478117
- PMCID: PMC11584403
- DOI: 10.1038/s43018-024-00773-6
Differential chromatin accessibility and transcriptional dynamics define breast cancer subtypes and their lineages
Abstract
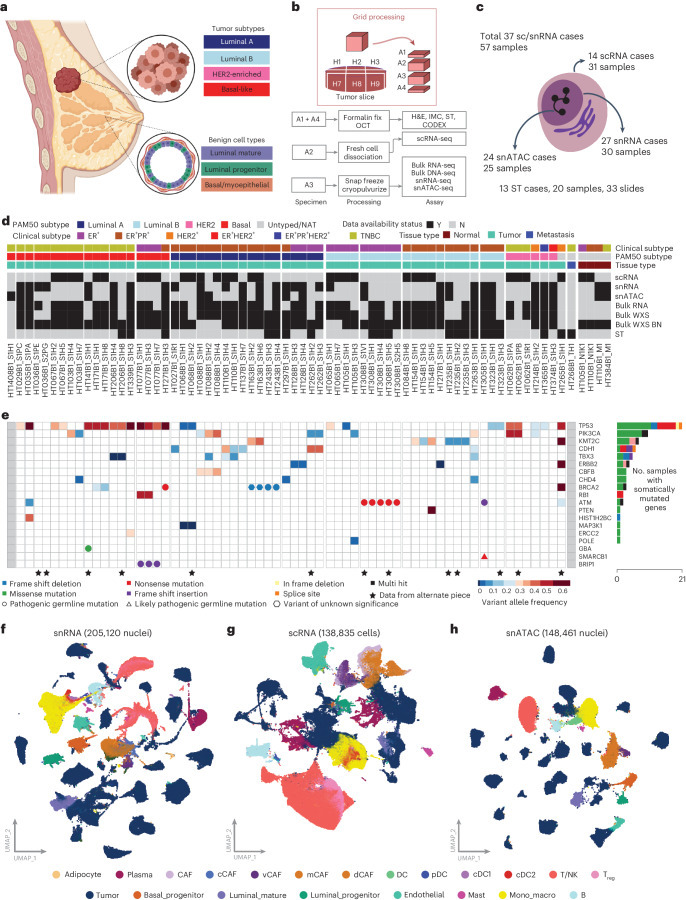
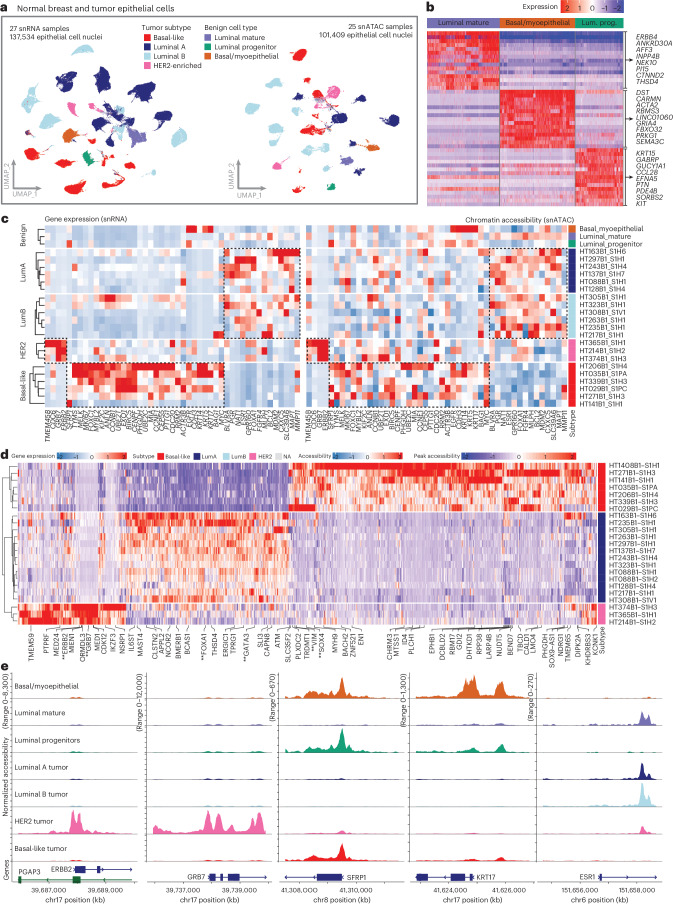
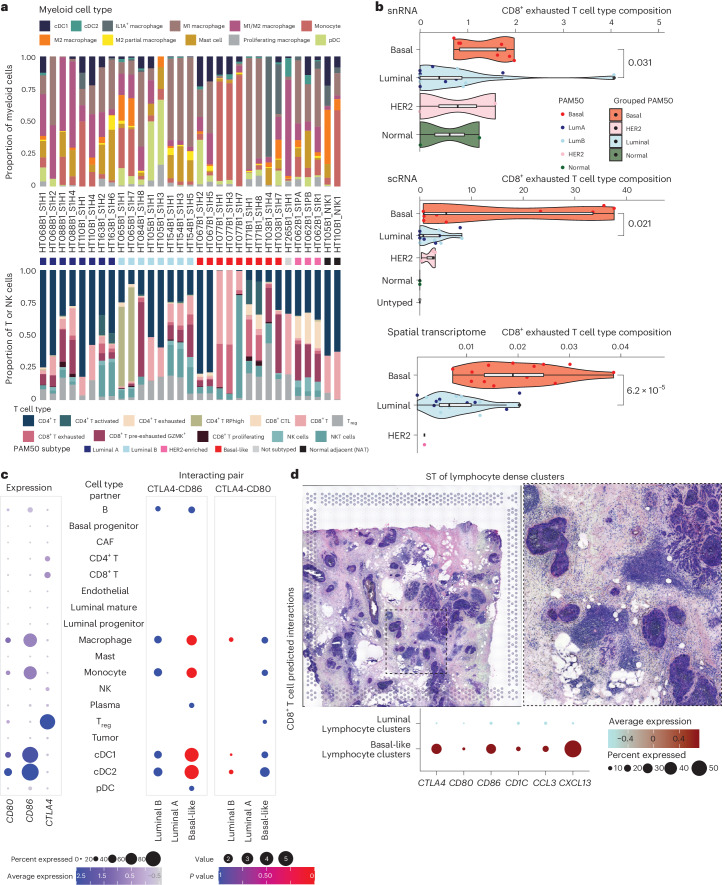
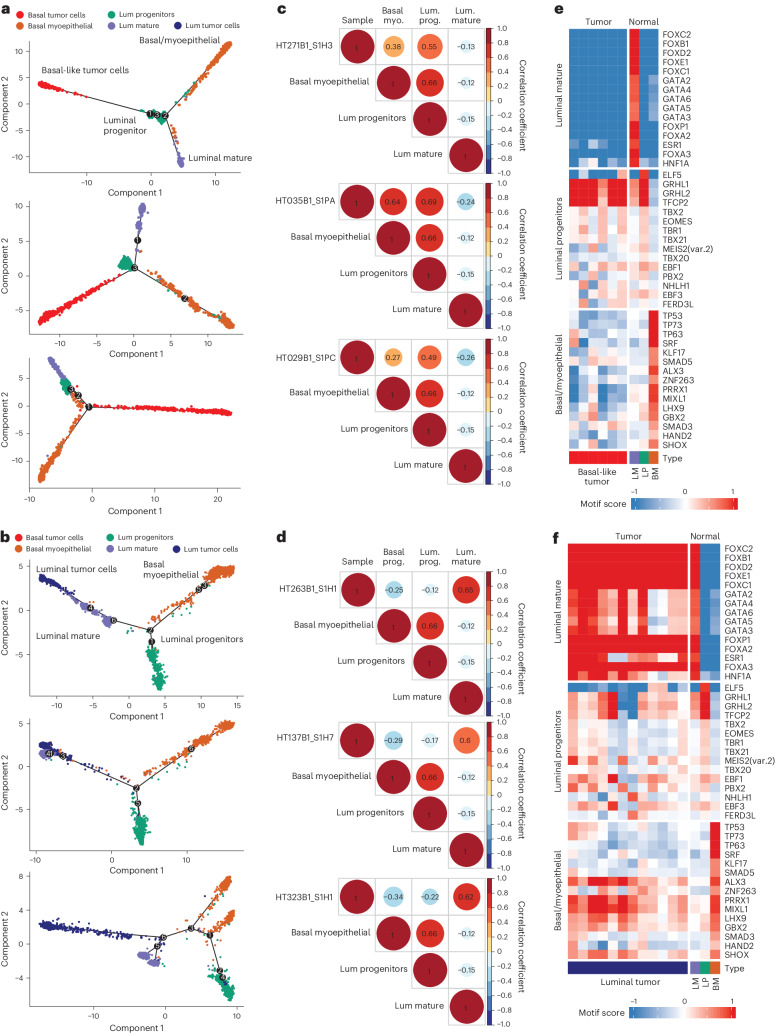
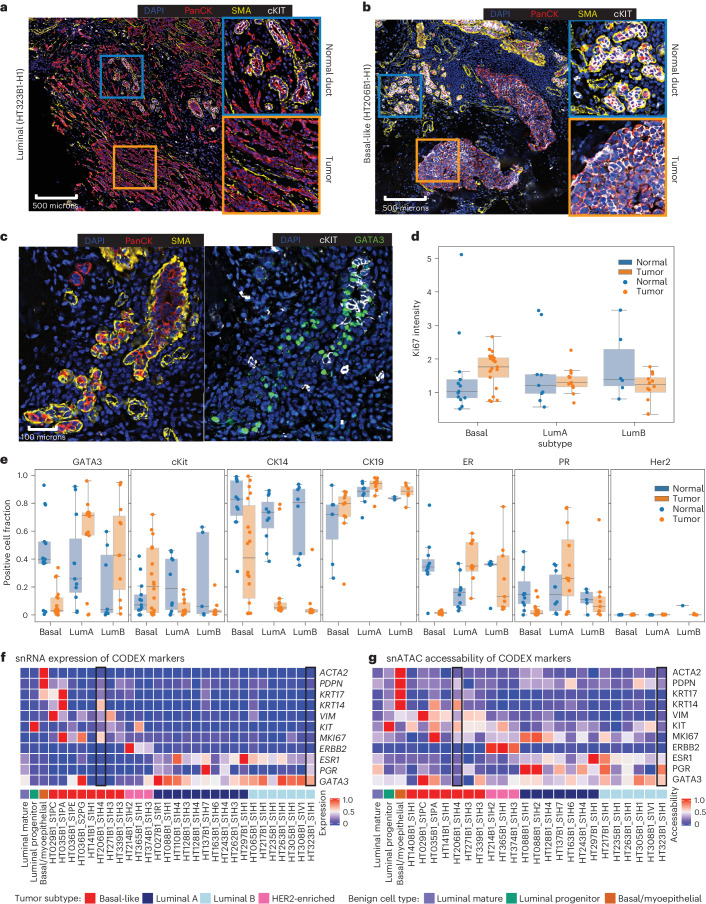
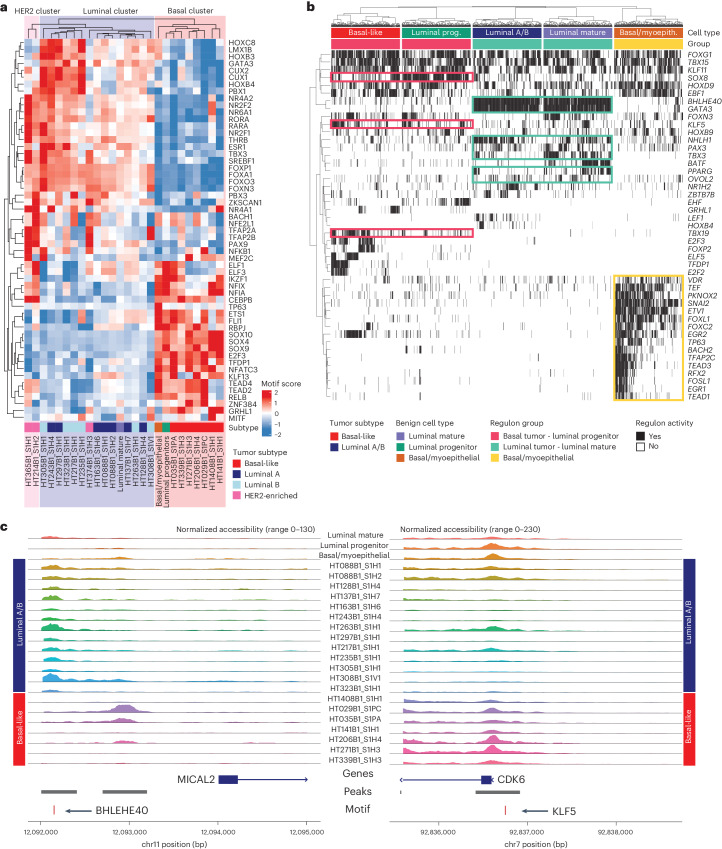
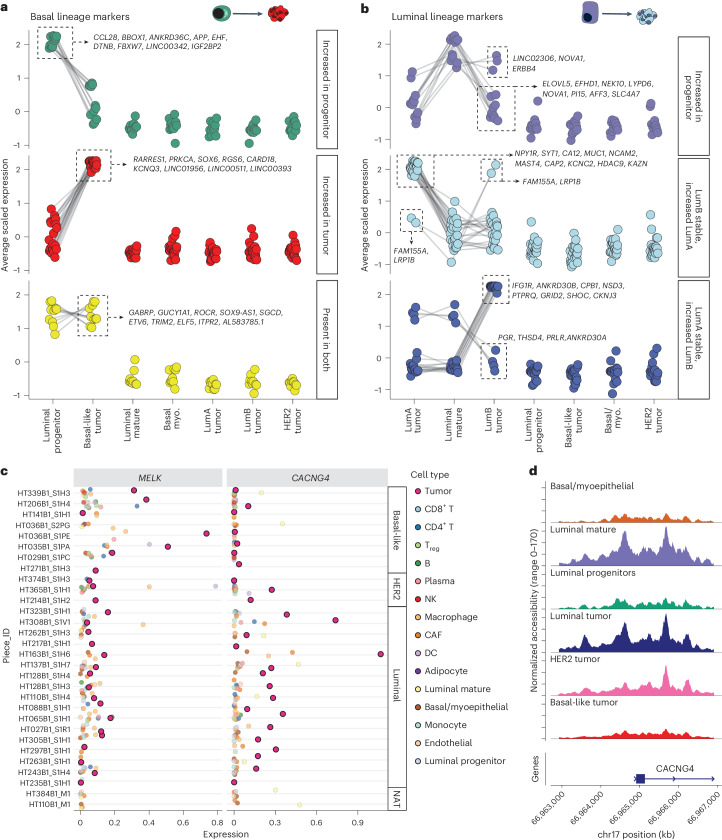
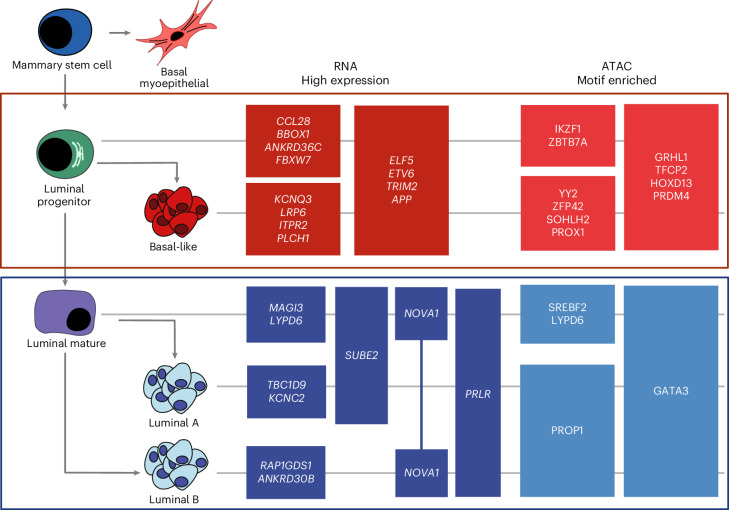
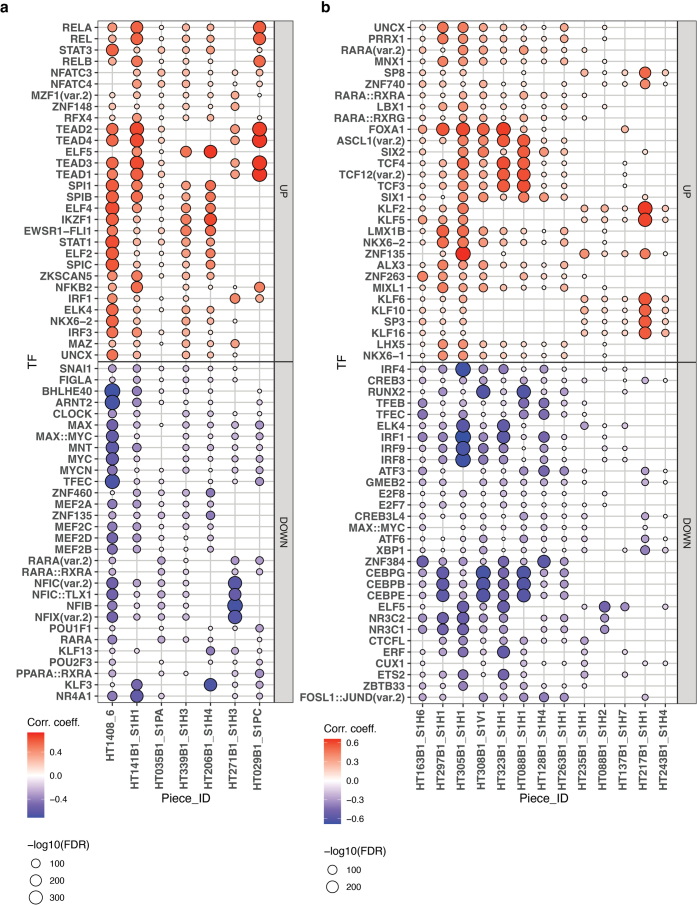
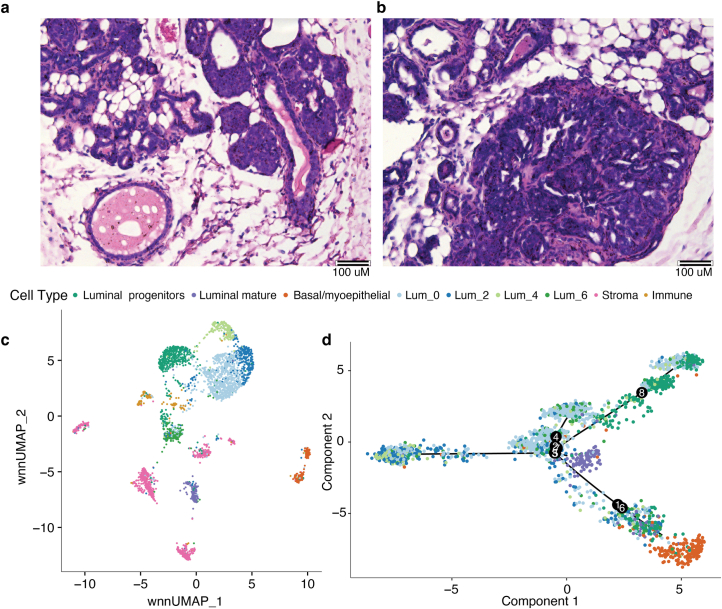
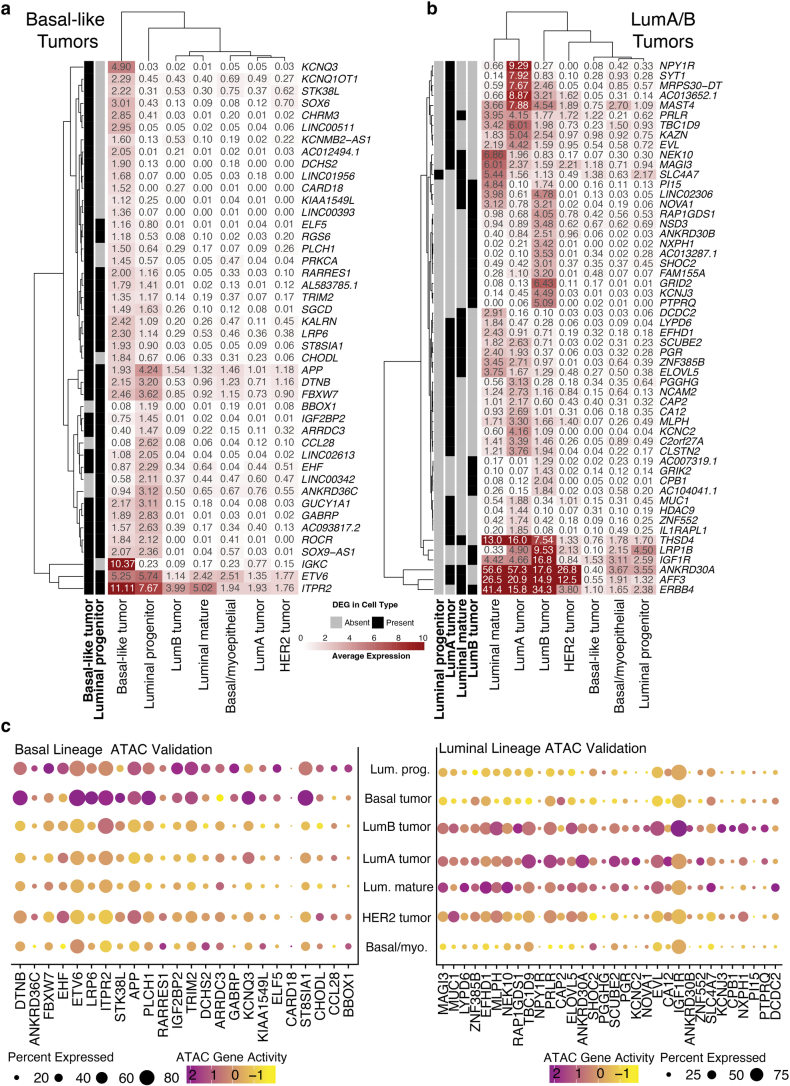
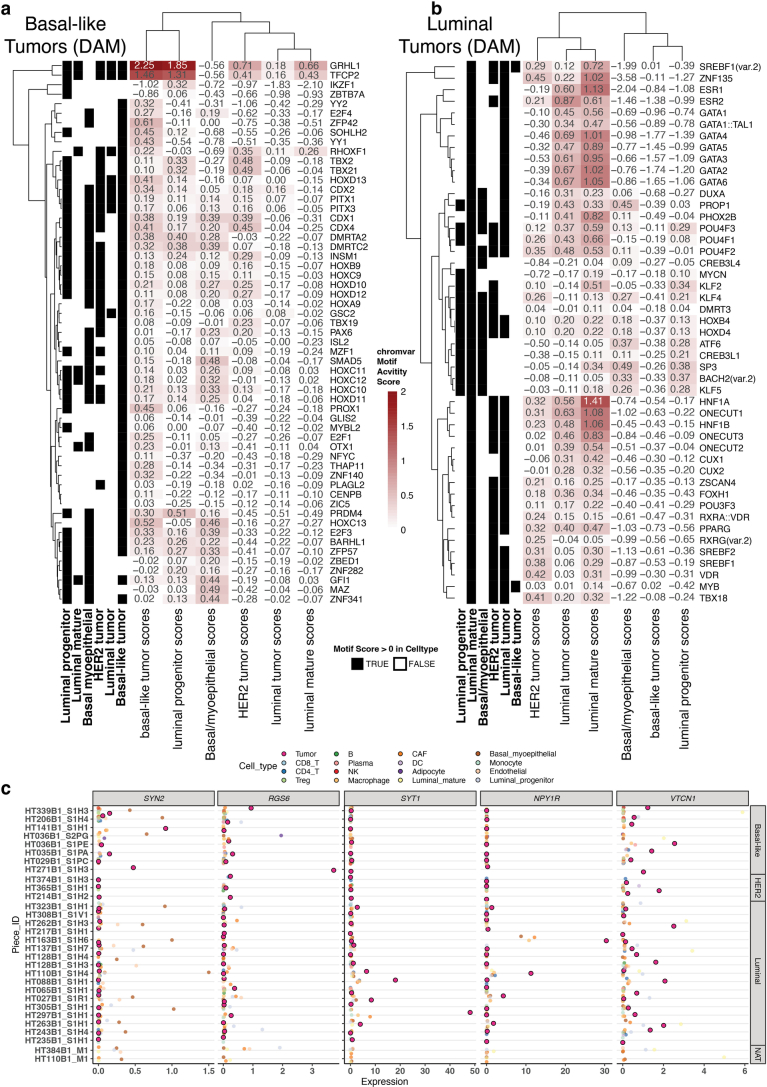
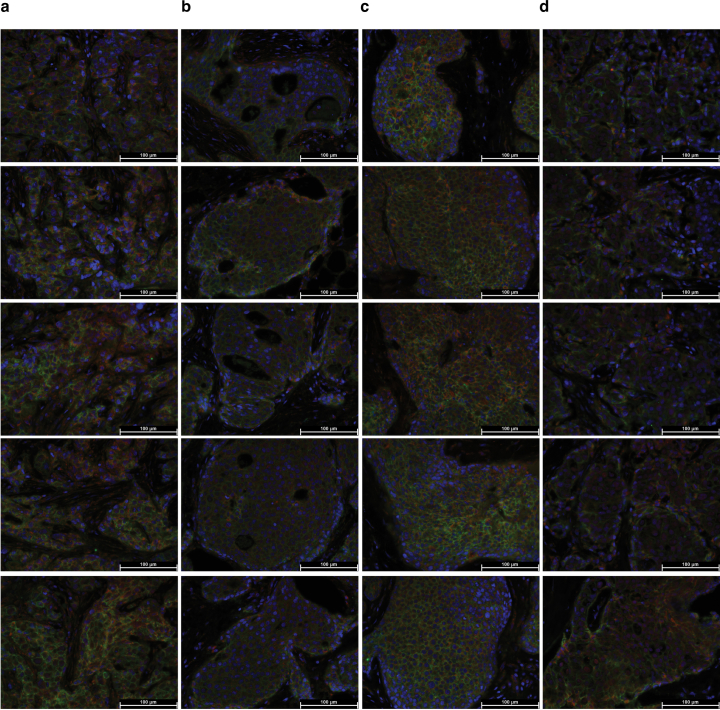
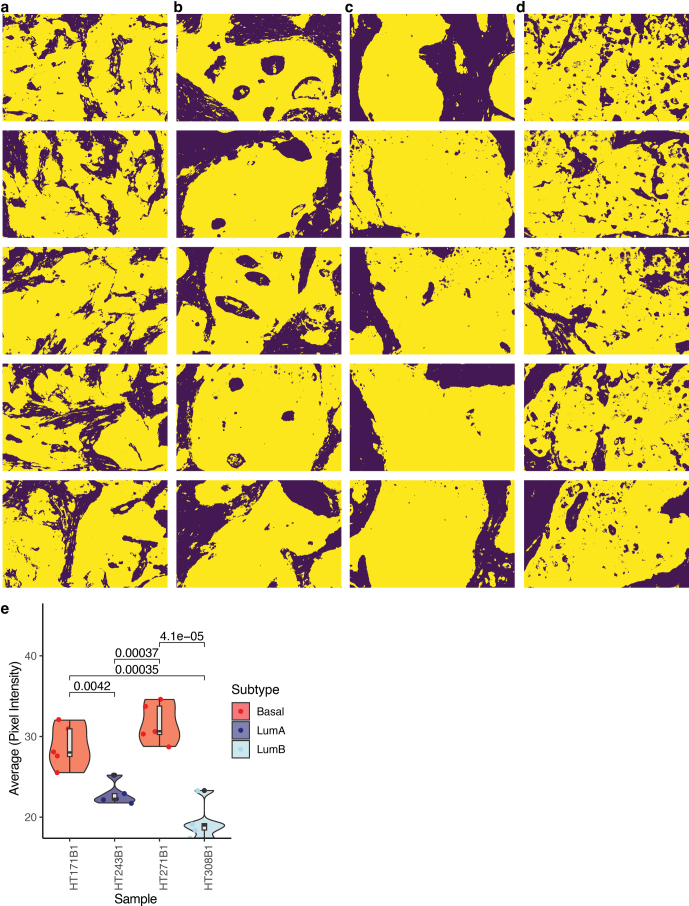
Breast cancer (BC) is defined by distinct molecular subtypes with different cells of origin. The transcriptional networks that characterize the subtype-specific tumor-normal lineages are not established. In this work, we applied bulk, single-cell and single-nucleus multi-omic techniques as well as spatial transcriptomics and multiplex imaging on 61 samples from 37 patients with BC to show characteristic links in gene expression and chromatin accessibility between BC subtypes and their putative cells of origin. Regulatory network analysis of transcription factors underscored the importance of BHLHE40 in luminal BC and luminal mature cells and KLF5 in basal-like tumors and luminal progenitor cells. Furthermore, we identify key genes defining the basal-like (SOX6 and KCNQ3) and luminal A/B (FAM155A and LRP1B) lineages. Exhausted CTLA4-expressing CD8+ T cells were enriched in basal-like BC, suggesting an altered means of immune dysfunction. These findings demonstrate analysis of paired transcription and chromatin accessibility at the single-cell level is a powerful tool for investigating cancer lineage and highlight transcriptional networks that define basal and luminal BC lineages.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Update of
-
Differential chromatin accessibility and transcriptional dynamics define breast cancer subtypes and their lineages.bioRxiv [Preprint]. 2023 Nov 2:2023.10.31.565031. doi: 10.1101/2023.10.31.565031. bioRxiv. 2023. Update in: Nat Cancer. 2024 Nov;5(11):1713-1736. doi: 10.1038/s43018-024-00773-6. PMID: 37961519 Free PMC article. Updated. Preprint.
References
-
- DeSantis, C. E. et al. Breast cancer statistics, 2019. CA Cancer J. Clin.69, 438–451 (2019). - PubMed
-
- Waks, A. G. & Winer, E. P. Breast cancer treatment: a review. JAMA321, 288–300 (2019). - PubMed
-
- Van Keymeulen, A. et al. Distinct stem cells contribute to mammary gland development and maintenance. Nature479, 189–193 (2011). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
