

Global loss of promoter-enhancer connectivity and rebalancing of gene expression during early colorectal cancer carcinogenesis
- PMID: 39478119
- PMCID: PMC11584406
- DOI: 10.1038/s43018-024-00823-z
Global loss of promoter-enhancer connectivity and rebalancing of gene expression during early colorectal cancer carcinogenesis
Erratum in
-
Author Correction: Global loss of promoter-enhancer connectivity and rebalancing of gene expression during early colorectal cancer carcinogenesis.Nat Cancer. 2025 Feb;6(2):404. doi: 10.1038/s43018-025-00915-4. Nat Cancer. 2025. PMID: 39865177 Free PMC article. No abstract available.
Abstract
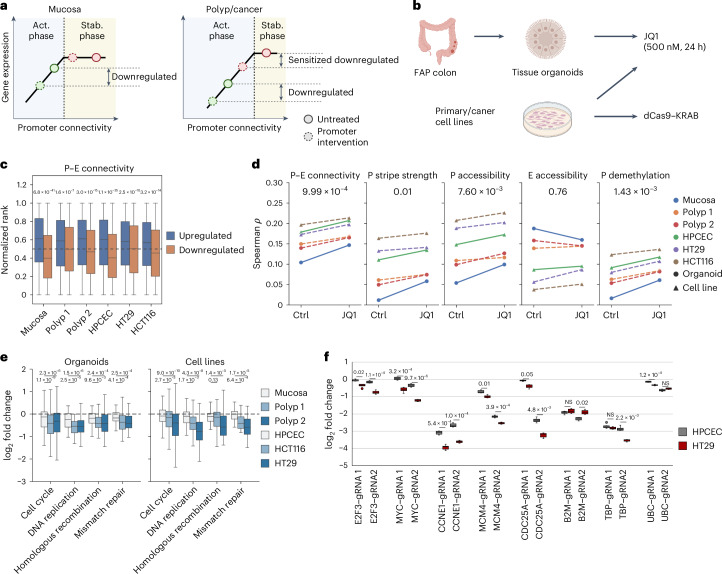
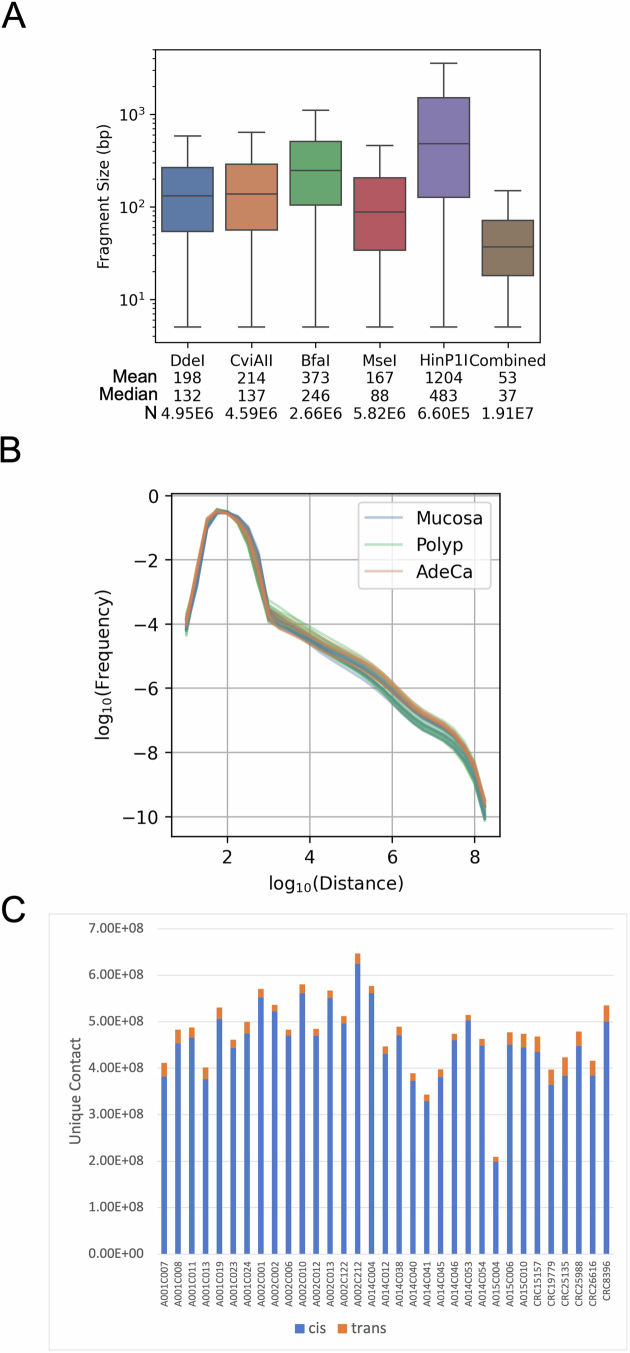
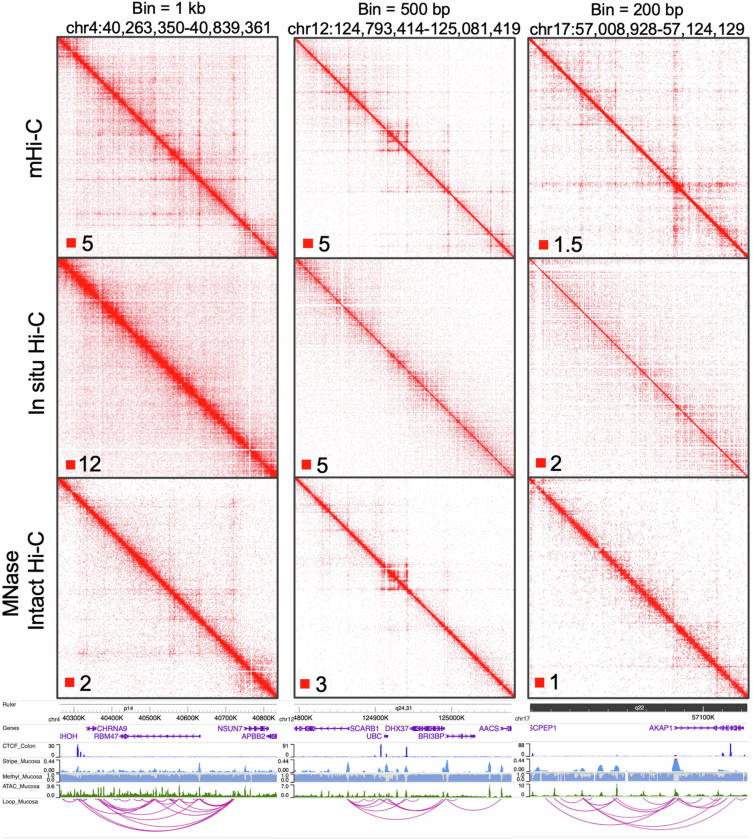
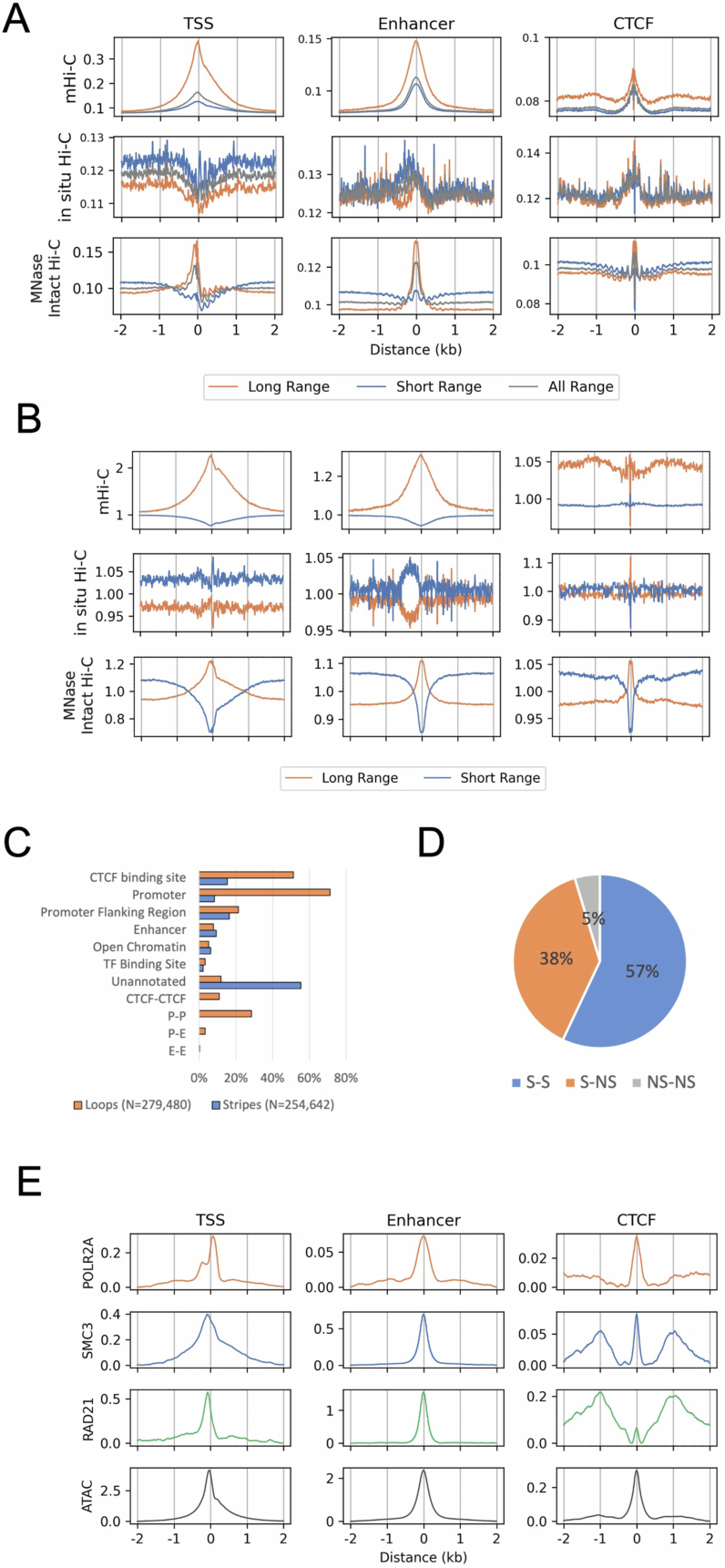
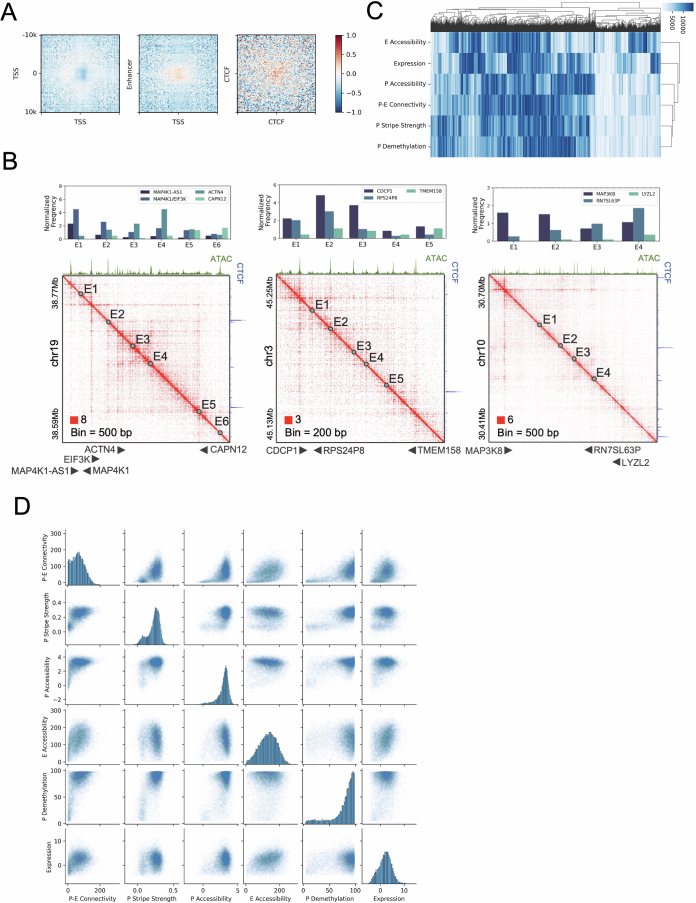
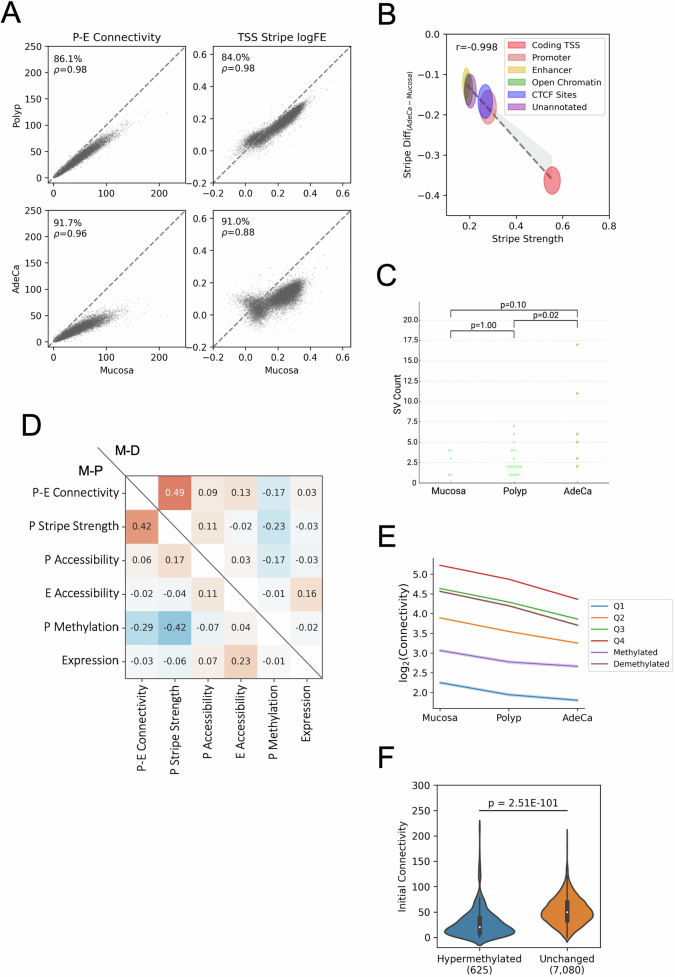
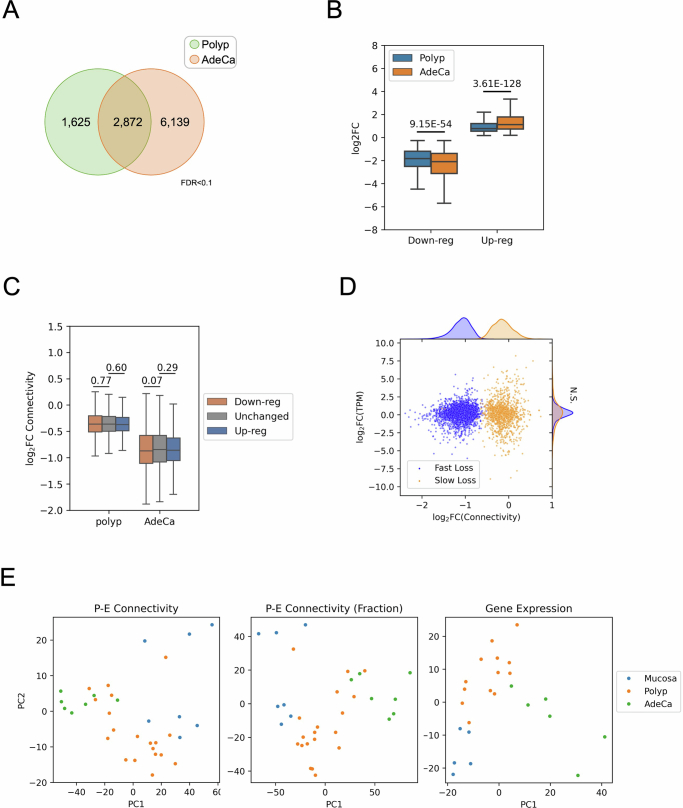
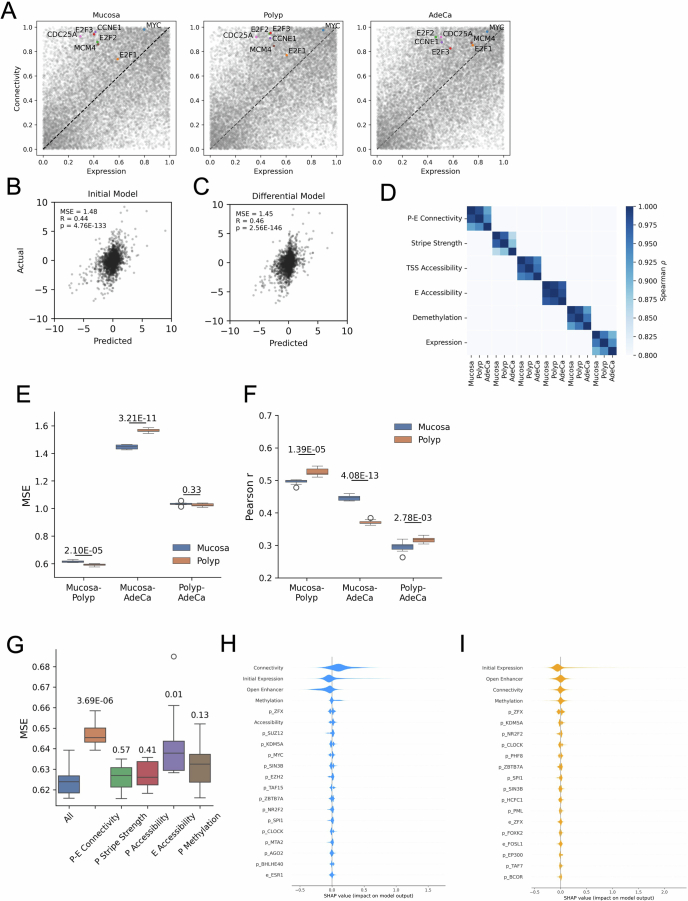
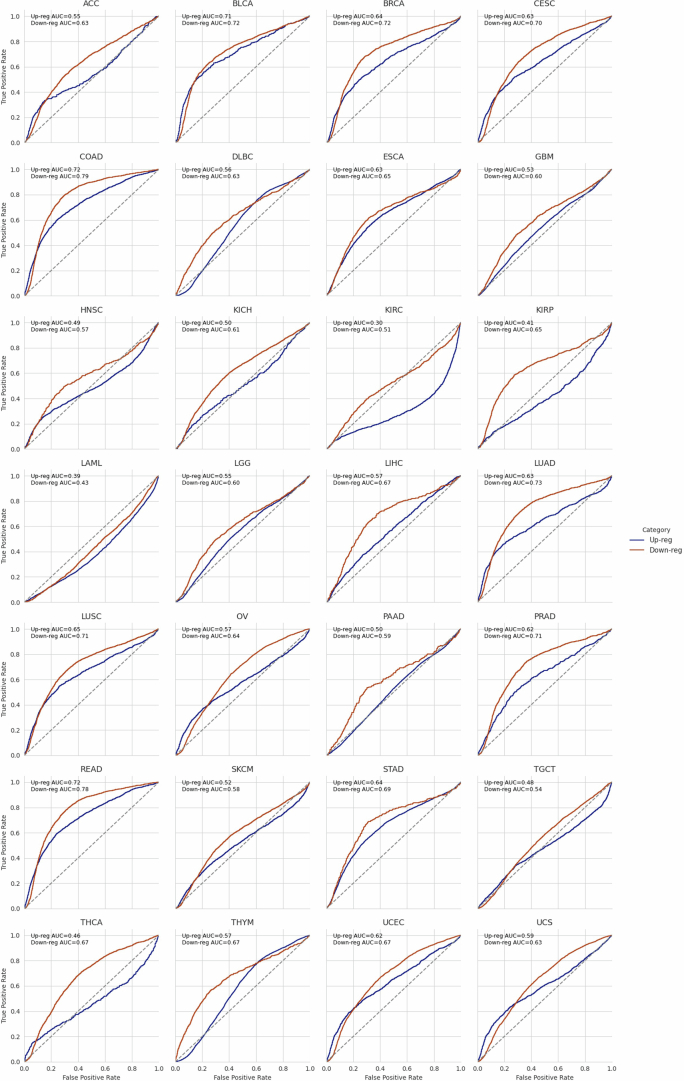
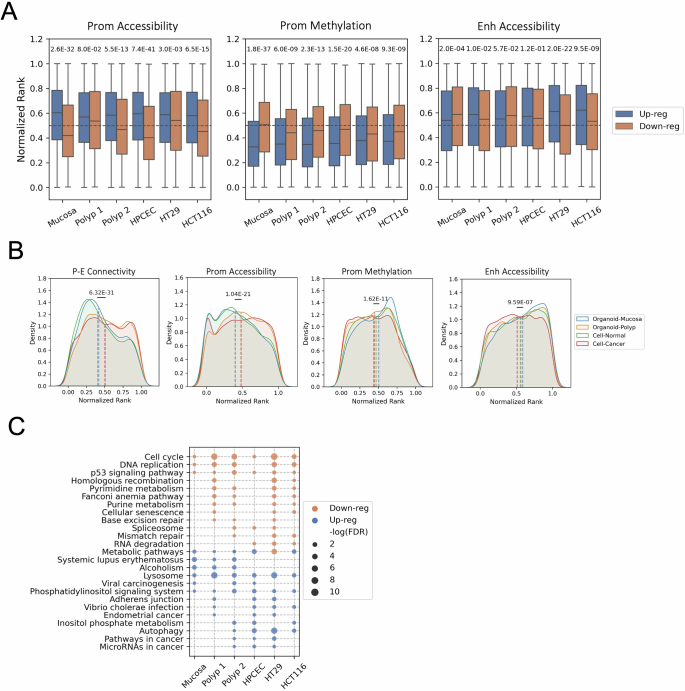
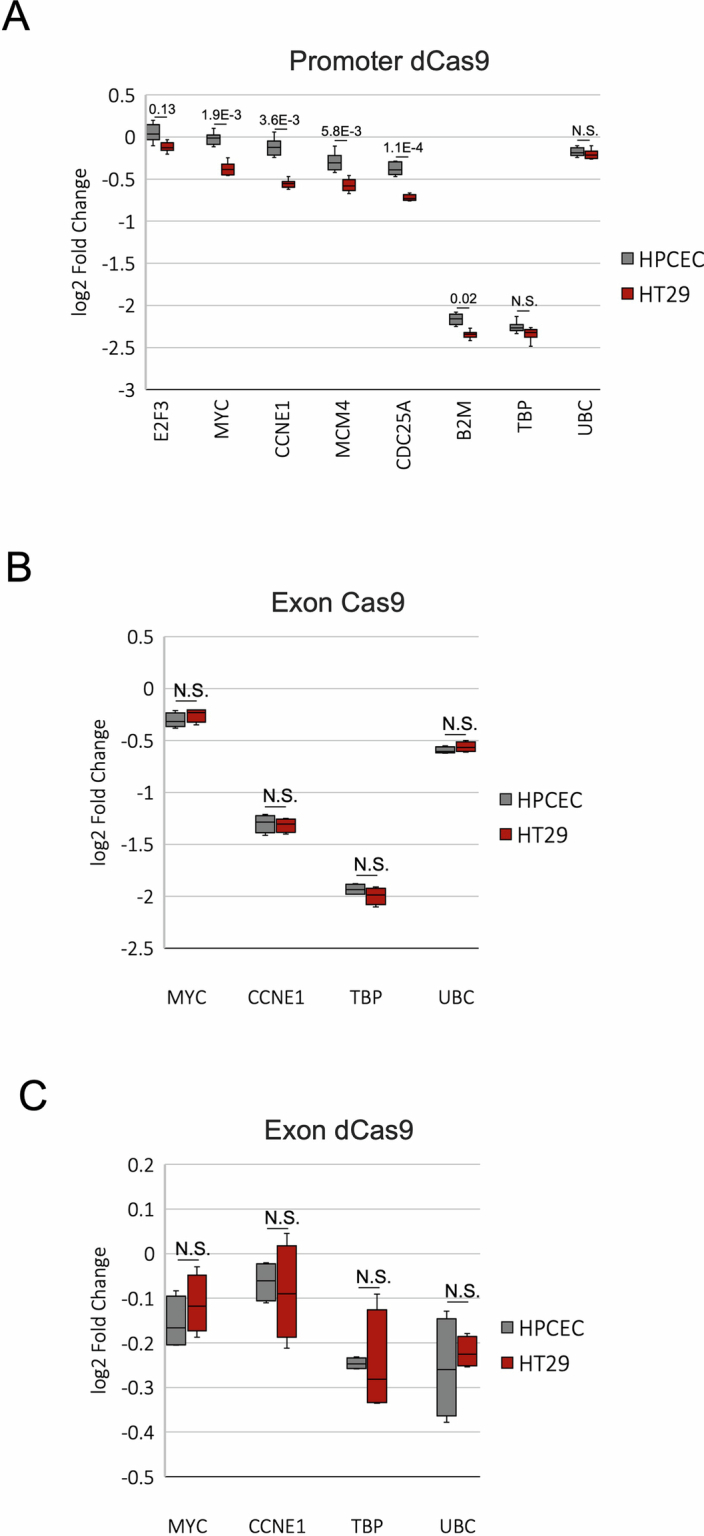
Although three-dimensional (3D) genome architecture is crucial for gene regulation, its role in disease remains elusive. We traced the evolution and malignant transformation of colorectal cancer (CRC) by generating high-resolution chromatin conformation maps of 33 colon samples spanning different stages of early neoplastic growth in persons with familial adenomatous polyposis (FAP). Our analysis revealed a substantial progressive loss of genome-wide cis-regulatory connectivity at early malignancy stages, correlating with nonlinear gene regulation effects. Genes with high promoter-enhancer (P-E) connectivity in unaffected mucosa were not linked to elevated baseline expression but tended to be upregulated in advanced stages. Inhibiting highly connected promoters preferentially represses gene expression in CRC cells compared to normal colonic epithelial cells. Our results suggest a two-phase model whereby neoplastic transformation reduces P-E connectivity from a redundant state to a rate-limiting one for transcriptional levels, highlighting the intricate interplay between 3D genome architecture and gene regulation during early CRC progression.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: G.K., A.J., D.L. and Z.S. are employees and shareholders of Ultima Genomics. M.P.S is a cofounder and scientific advisor of Personalis, Qbio, SensOmics, January AI, Mirvie, Protos, NiMo and Onza and is on the advisory board of Genapsys. E.D.E. is an employee and stockholder of Invitae and an advisor and stockholder of Taproot Health and Exir Bio. W.J.G. has affiliations with Guardant Health (consultant and scientific advisory board), Protillion Biosciences (Scientifica cofounder) and 10x and has licensed patents associated with ATAC-seq. All other authors declare no competing interests.
Figures
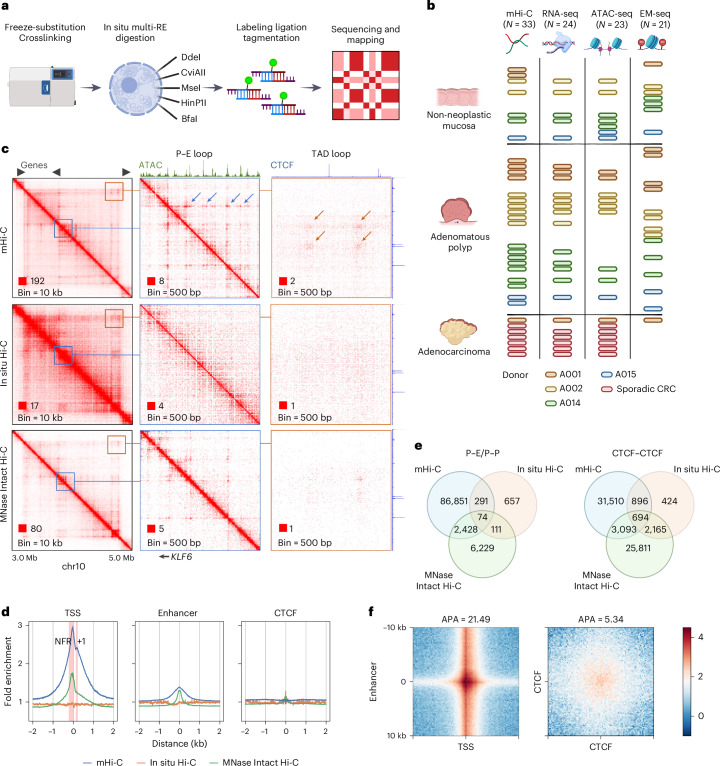
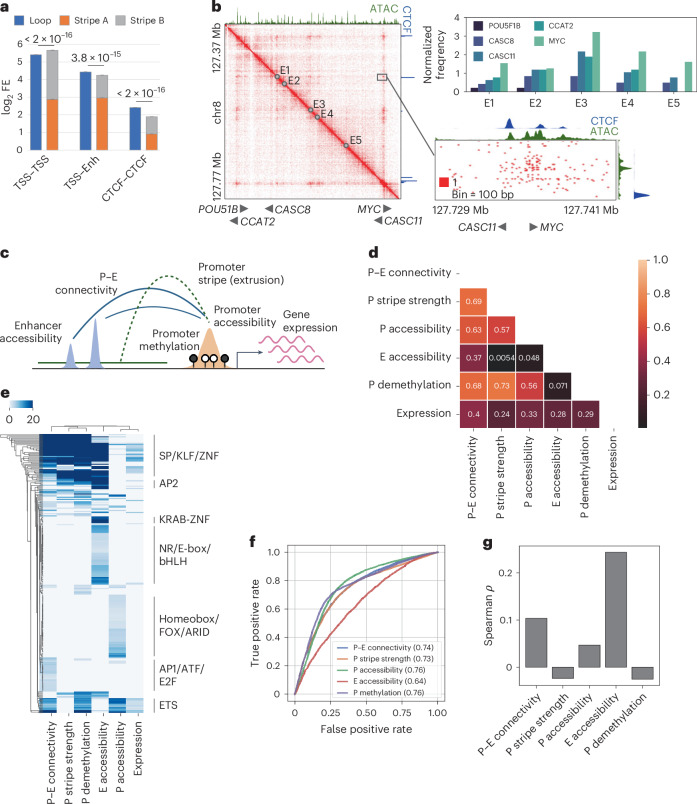
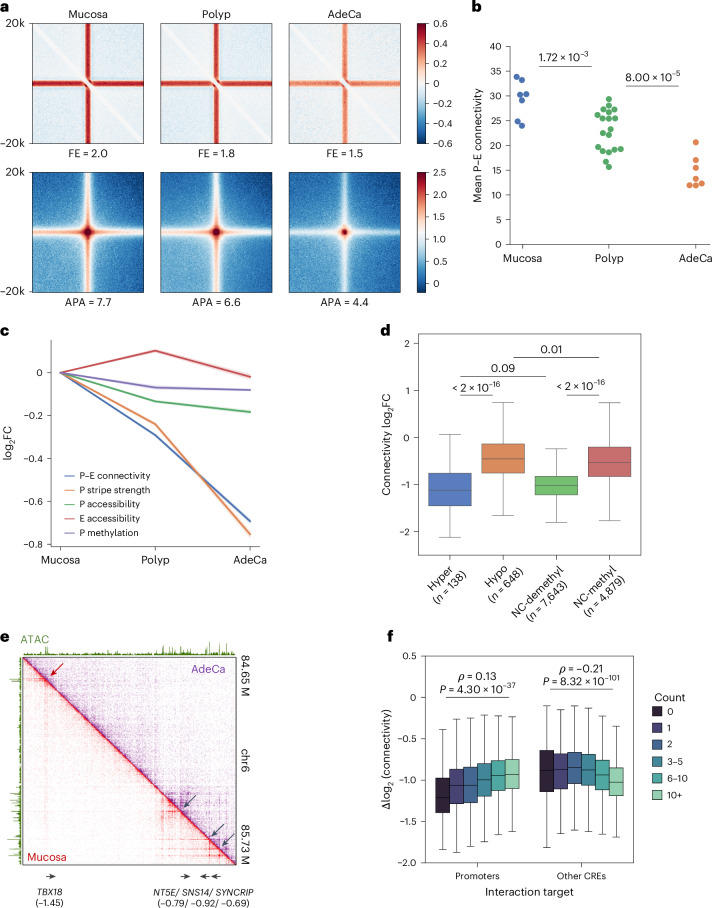
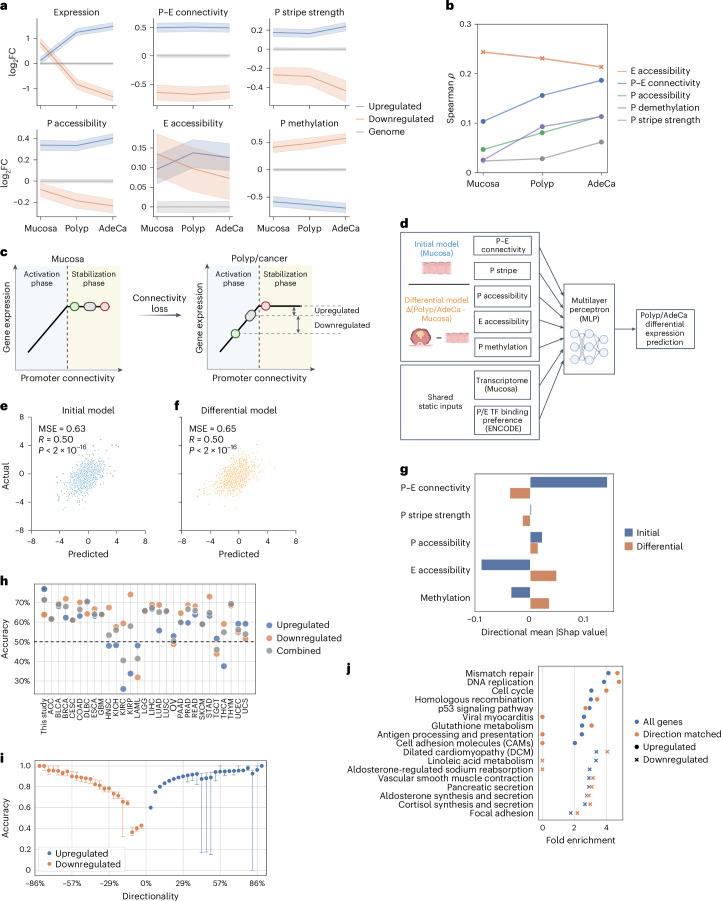
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
