

A spatial hierarchical network learning framework for drug repositioning allowing interpretation from macro to micro scale
- PMID: 39478146
- PMCID: PMC11525566
- DOI: 10.1038/s42003-024-07107-3
A spatial hierarchical network learning framework for drug repositioning allowing interpretation from macro to micro scale
Abstract
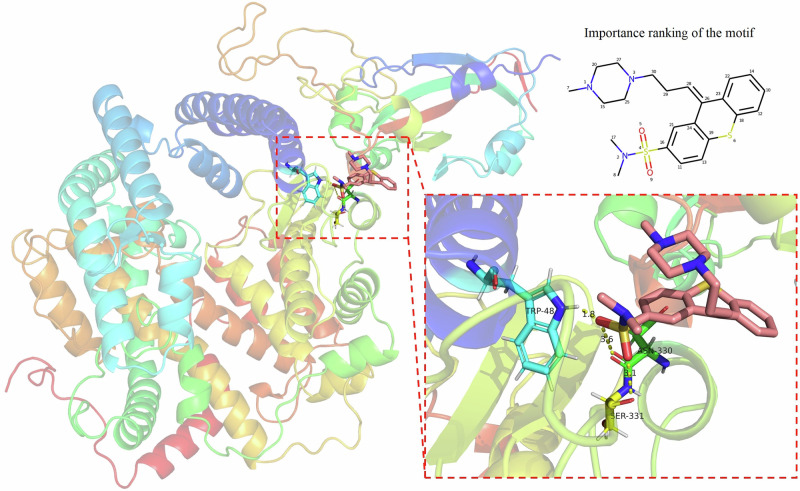
Biomedical network learning offers fresh prospects for expediting drug repositioning. However, traditional network architectures struggle to quantify the relationship between micro-scale drug spatial structures and corresponding macro-scale biomedical networks, limiting their ability to capture key pharmacological properties and complex biomedical information crucial for drug screening and therapeutic discovery. Moreover, challenges such as difficulty in capturing long-range dependencies hinder current network-based approaches. To address these limitations, we introduce the Spatial Hierarchical Network, modeling molecular 3D structures and biological associations into a unified network. We propose an end-to-end framework, SpHN-VDA, integrating spatial hierarchical information through triple attention mechanisms to enhance machine understanding of molecular functionality and improve the accuracy of virus-drug association identification. SpHN-VDA outperforms leading models across three datasets, particularly excelling in out-of-distribution and cold-start scenarios. It also exhibits enhanced robustness against data perturbation, ranging from 20% to 40%. It accurately identifies critical motifs for binding sites, even without protein residue annotations. Leveraging reliability of SpHN-VDA, we have identified 25 potential candidate drugs through gene expression analysis and CMap. Molecular docking experiments with the SARS-CoV-2 spike protein further corroborate the predictions. This research highlights the broad potential of SpHN-VDA to enhance drug repositioning and identify effective treatments for various diseases.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
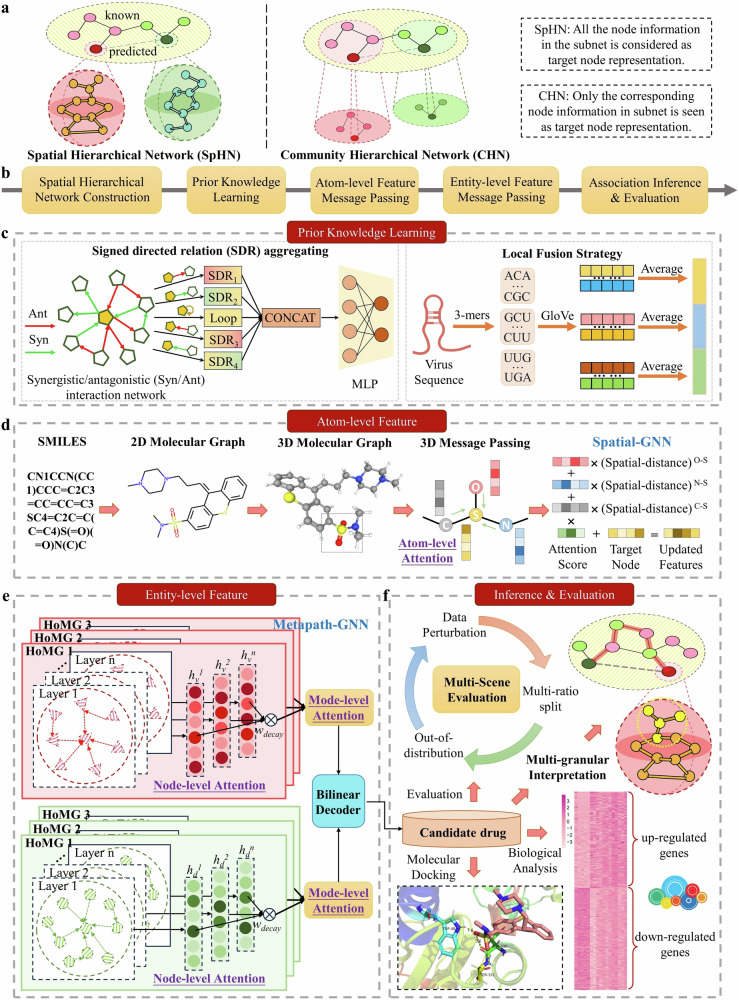
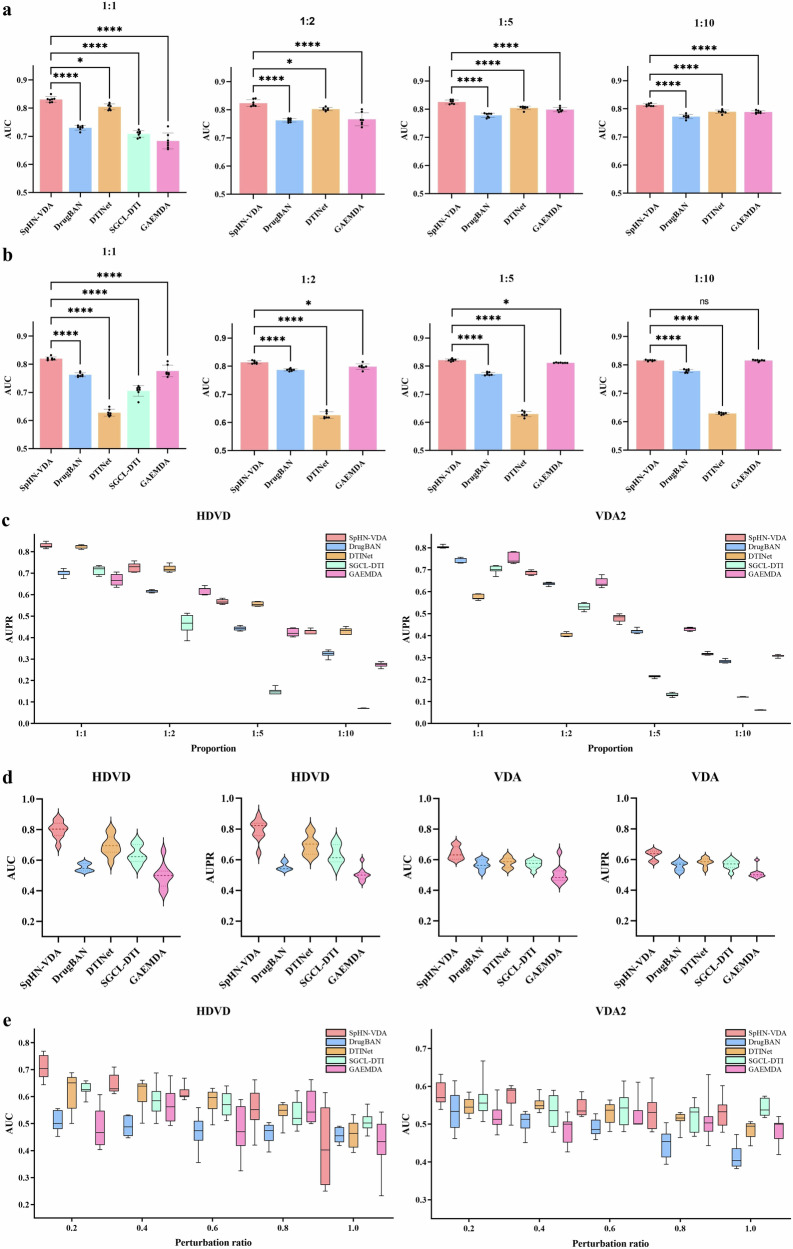

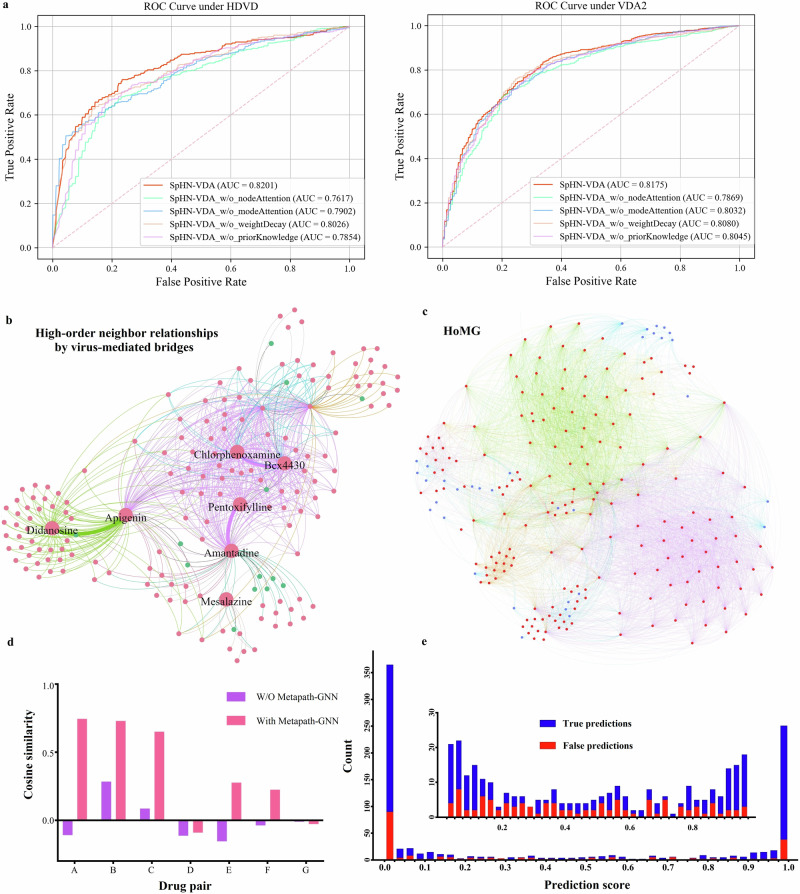
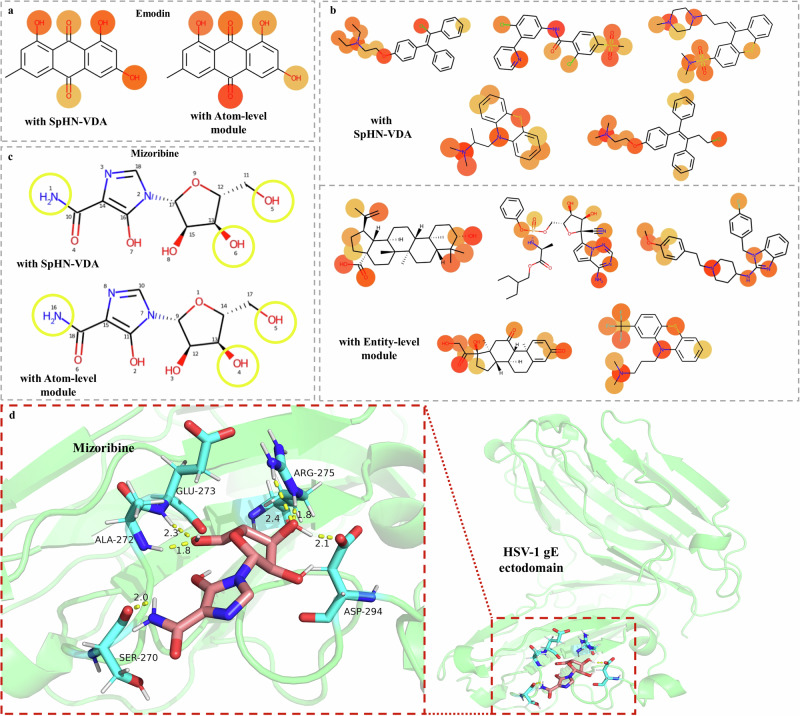
References
-
- Dickson, M. & Gagnon, J. P. Key factors in the rising cost of new drug discovery and development. Nat. Rev. Drug Discov.3, 417–429 (2004). - PubMed
-
- Pushpakom, S. et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov.18, 41–58 (2019). - PubMed
-
- Guy, R. K., DiPaola, R. S., Romanelli, F. & Dutch, R. E. Rapid repurposing of drugs for COVID-19. Sci368, 829–830 (2020). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
