

Polyclonality overcomes fitness barriers in Apc-driven tumorigenesis
- PMID: 39478206
- PMCID: PMC11525183
- DOI: 10.1038/s41586-024-08053-0
Polyclonality overcomes fitness barriers in Apc-driven tumorigenesis
Abstract
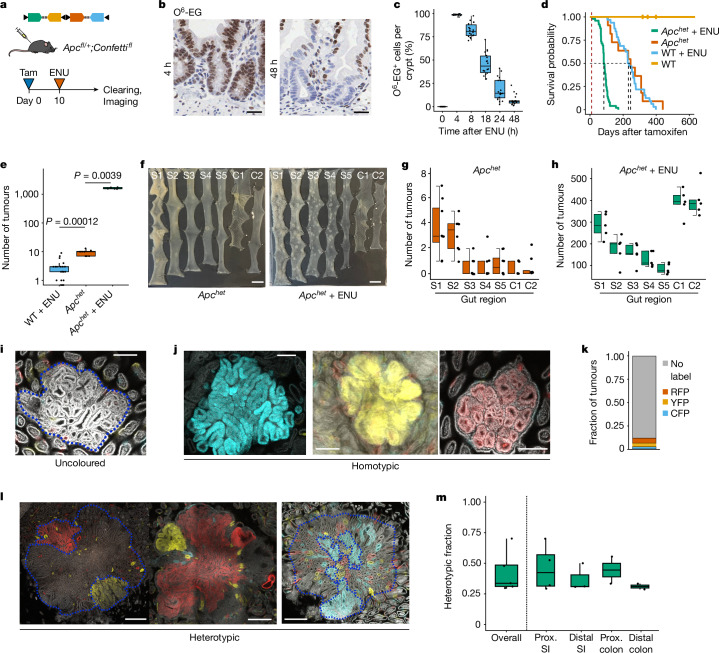
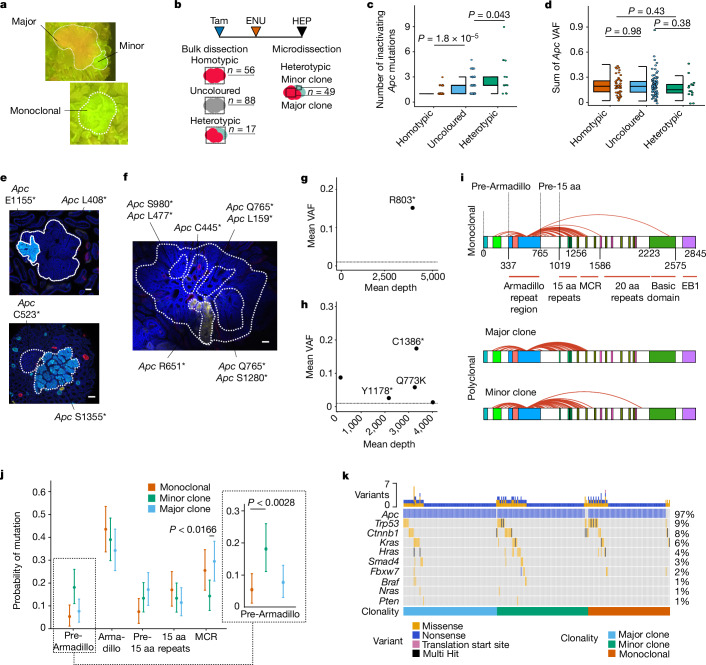
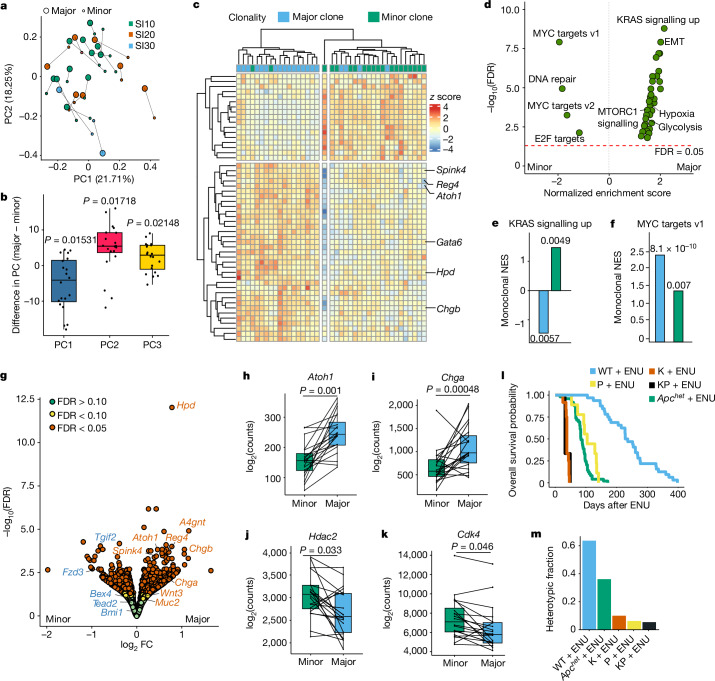
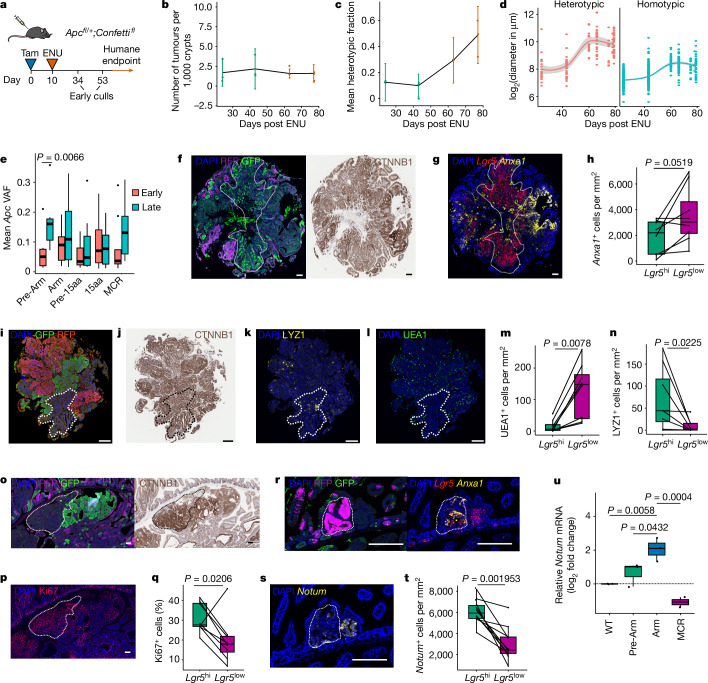
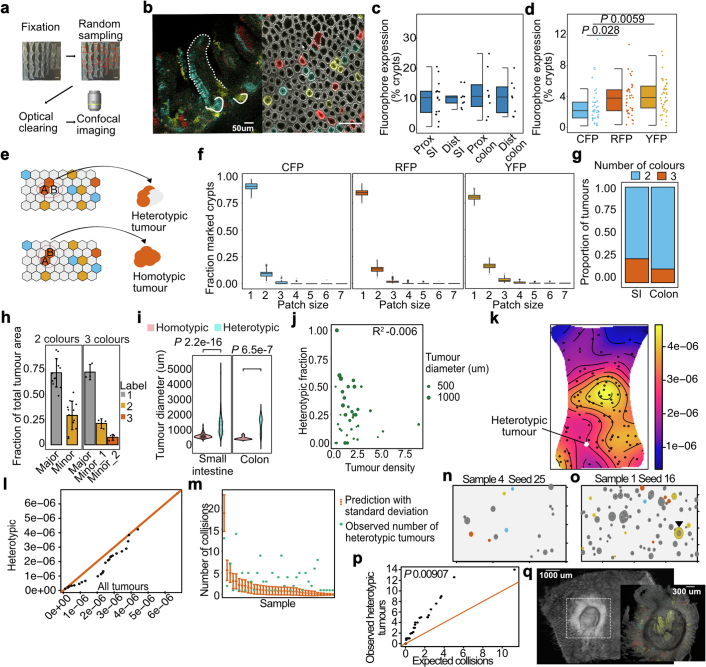
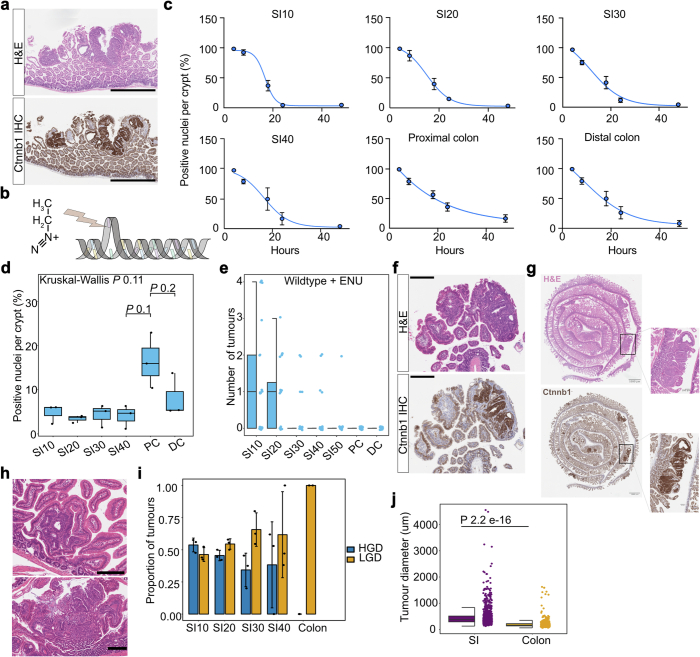
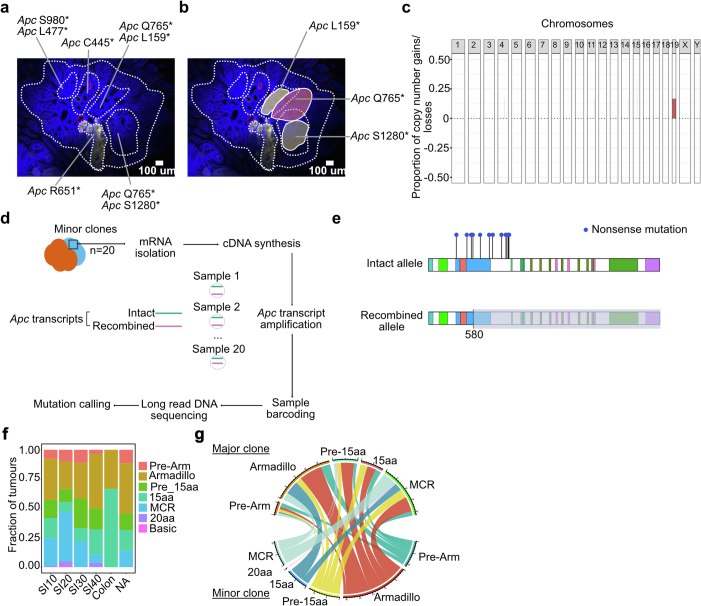
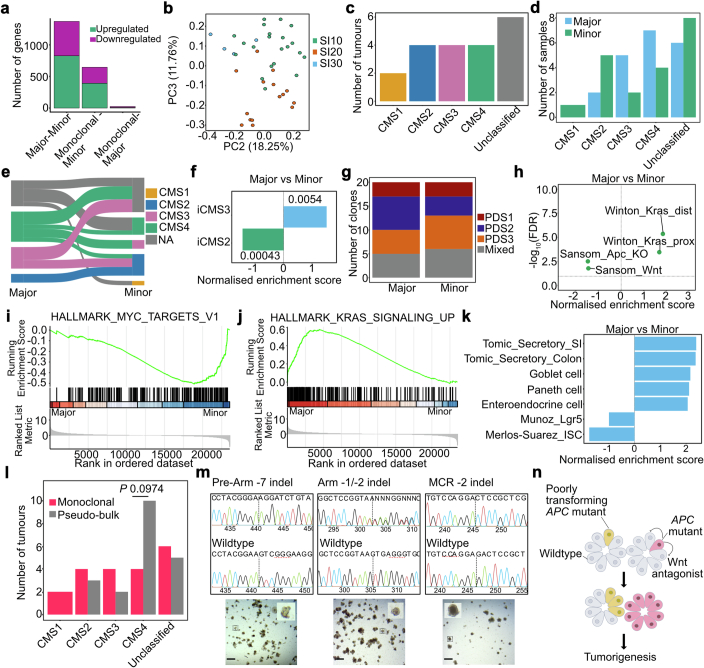
Loss-of-function mutations in the tumour suppressor APC are an initial step in intestinal tumorigenesis1,2. APC-mutant intestinal stem cells outcompete their wild-type neighbours through the secretion of Wnt antagonists, which accelerates the fixation and subsequent rapid clonal expansion of mutants3-5. Reports of polyclonal intestinal tumours in human patients and mouse models appear at odds with this process6,7. Here we combine multicolour lineage tracing with chemical mutagenesis in mice to show that a large proportion of intestinal tumours have a multiancestral origin. Polyclonal tumours retain a structure comprising subclones with distinct Apc mutations and transcriptional states, driven predominantly by differences in KRAS and MYC signalling. These pathway-level changes are accompanied by profound differences in cancer stem cell phenotypes. Of note, these findings are confirmed by introducing an oncogenic Kras mutation that results in predominantly monoclonal tumour formation. Further, polyclonal tumours have accelerated growth dynamics, suggesting a link between polyclonality and tumour progression. Together, these findings demonstrate the role of interclonal interactions in promoting tumorigenesis through non-cell autonomous pathways that are dependent on the differential activation of oncogenic pathways between clones.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Fearon, E. R. & Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell61, 759–767 (1990). - PubMed
-
- Barker, N. et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature457, 608–611 (2009). - PubMed
-
- Van Neerven, S. M. et al. Apc-mutant cells act as supercompetitors in intestinal tumour initiation. Nature594, 436–441 (2021). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
