

Temporal recording of mammalian development and precancer
- PMID: 39478207
- PMCID: PMC11525190
- DOI: 10.1038/s41586-024-07954-4
Temporal recording of mammalian development and precancer
Abstract
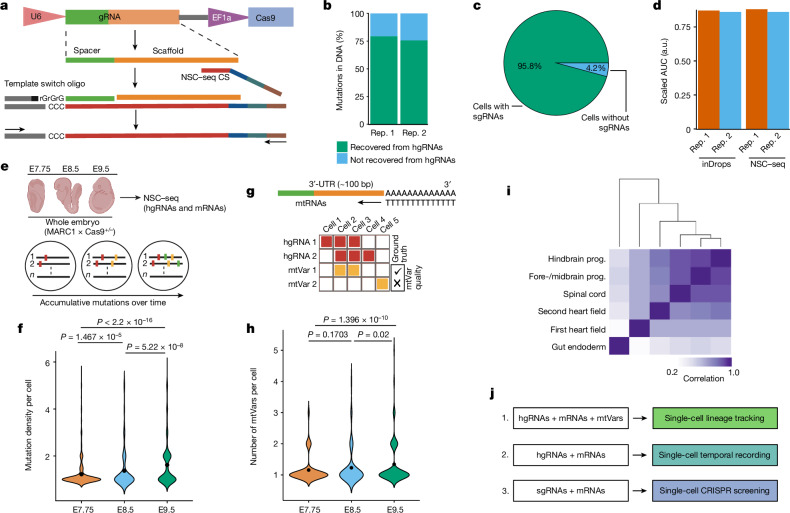
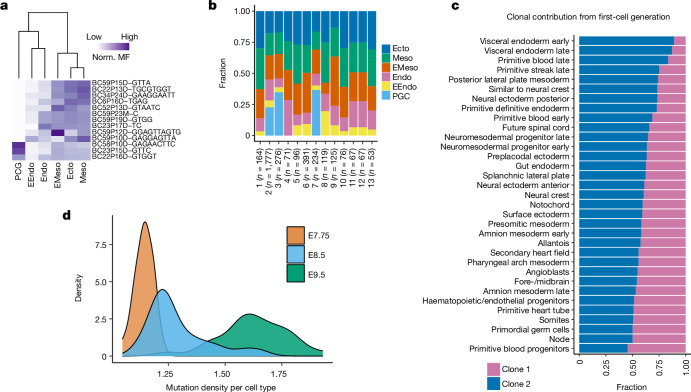
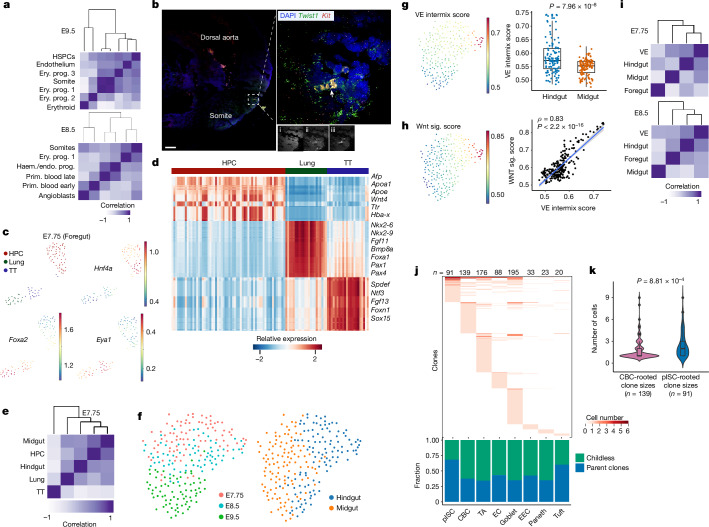
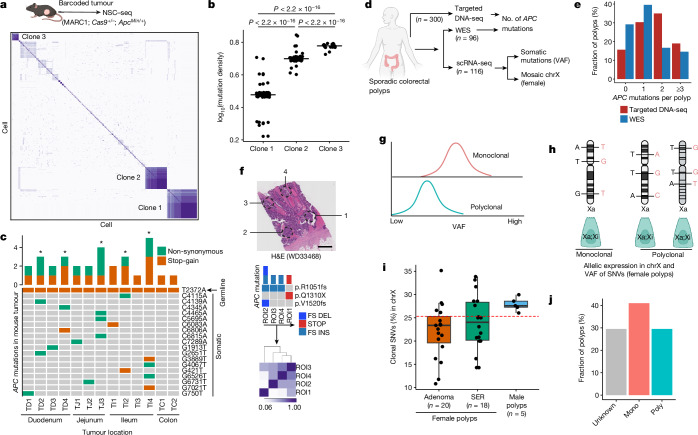
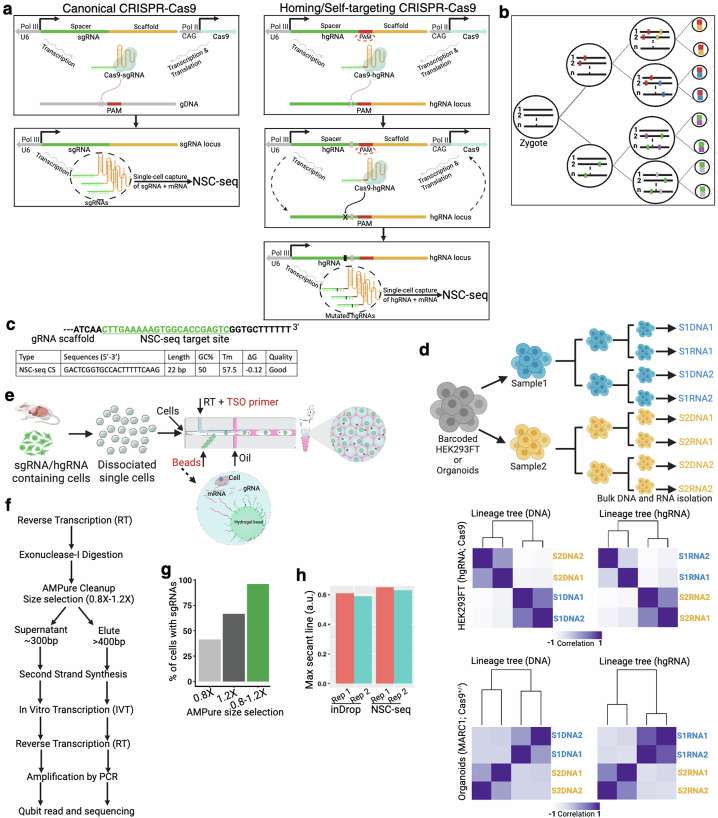
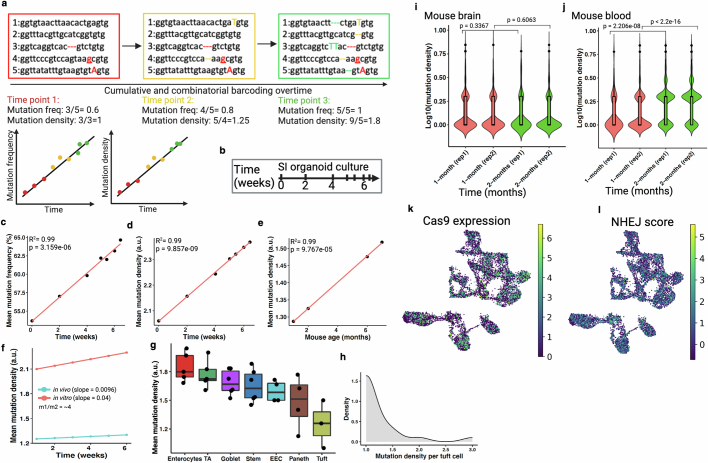
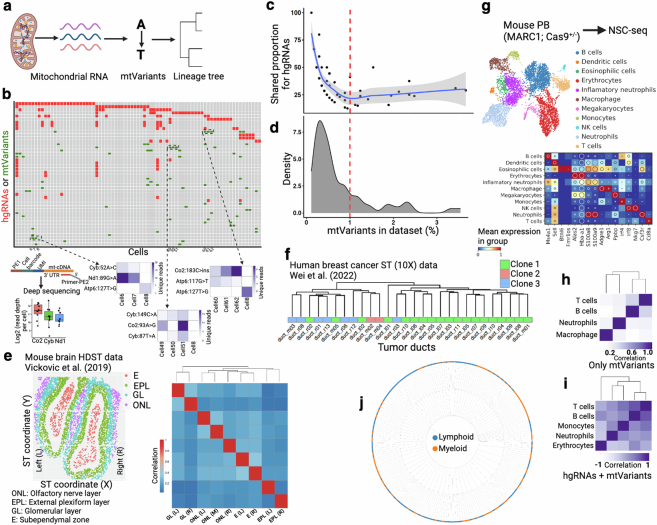
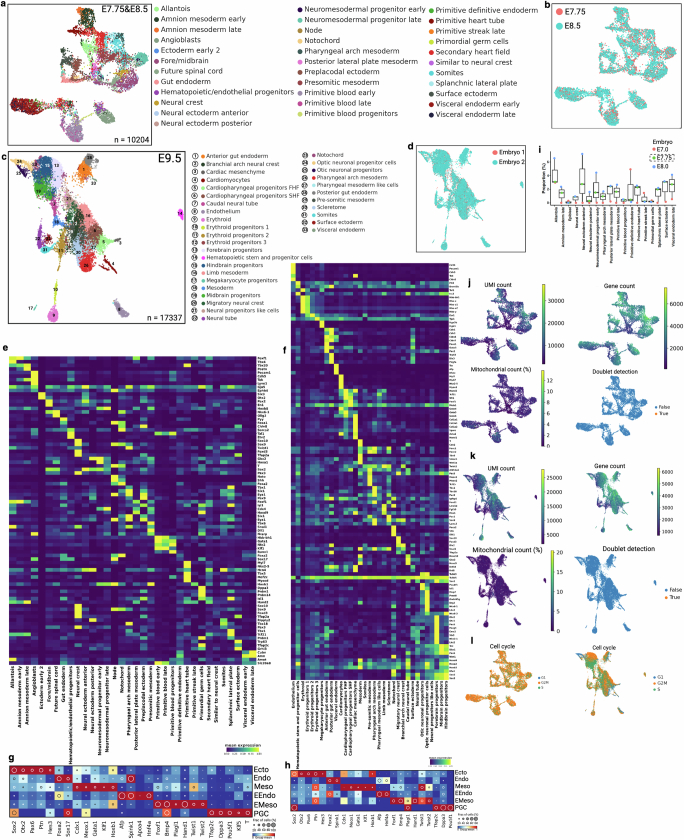
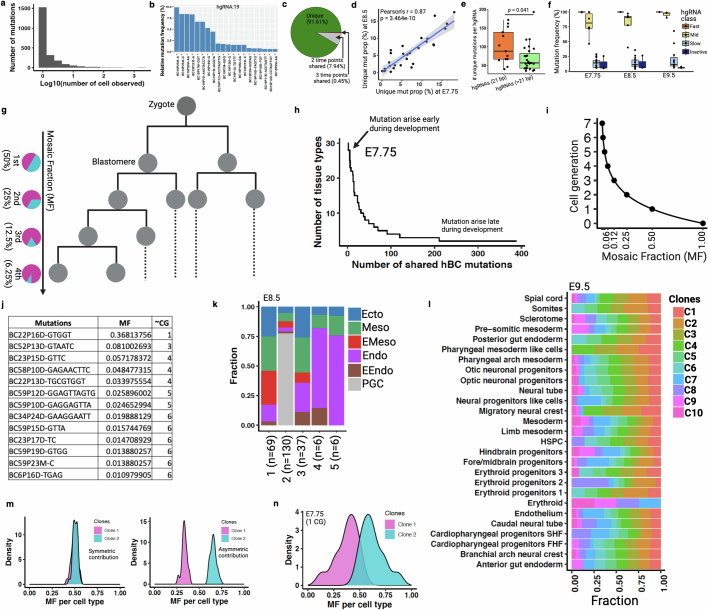
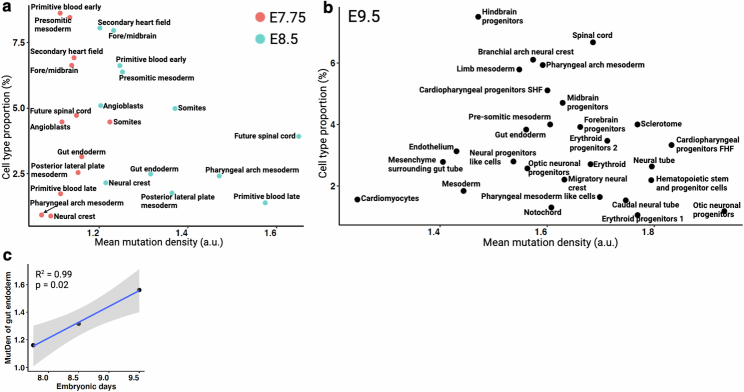
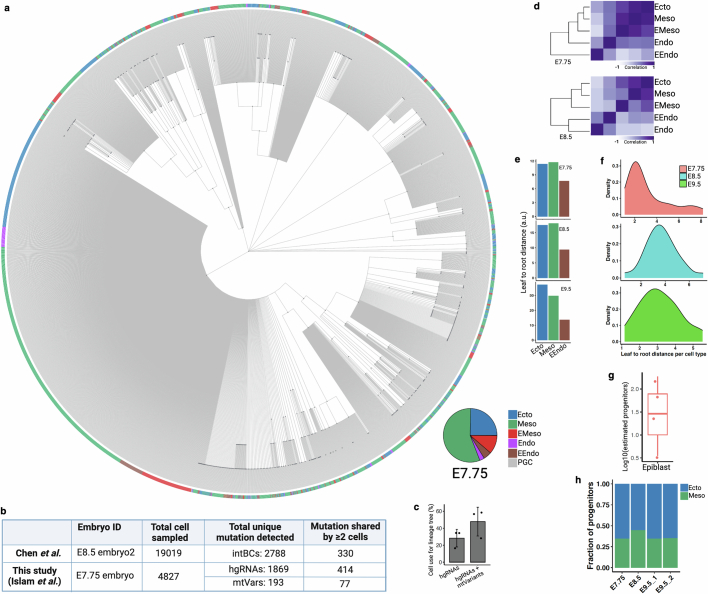
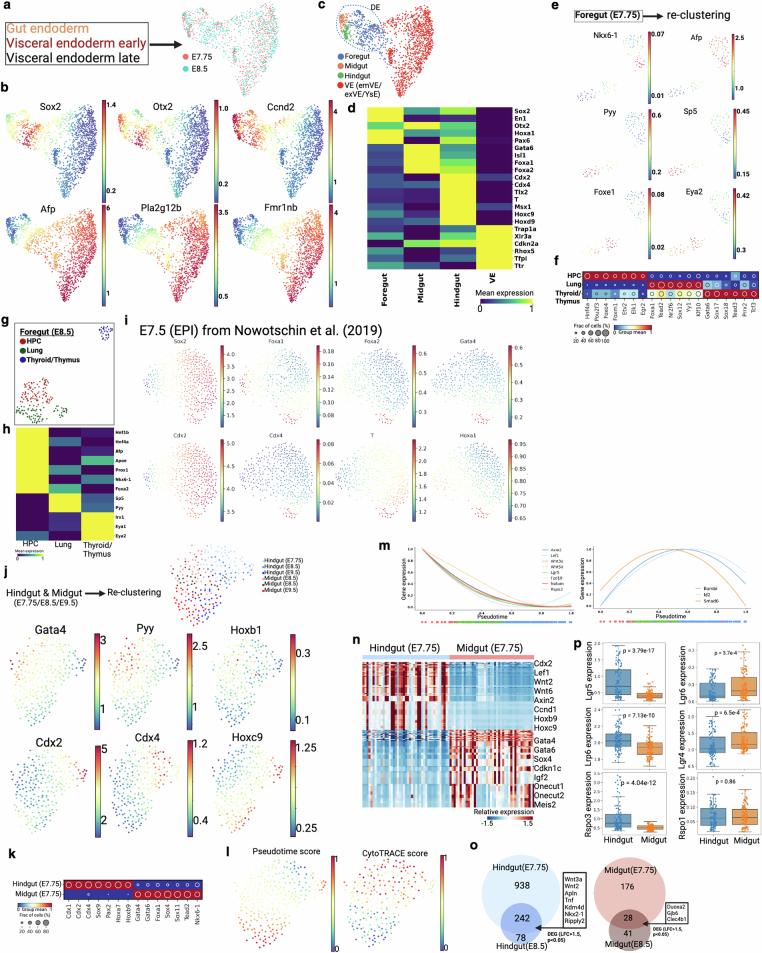
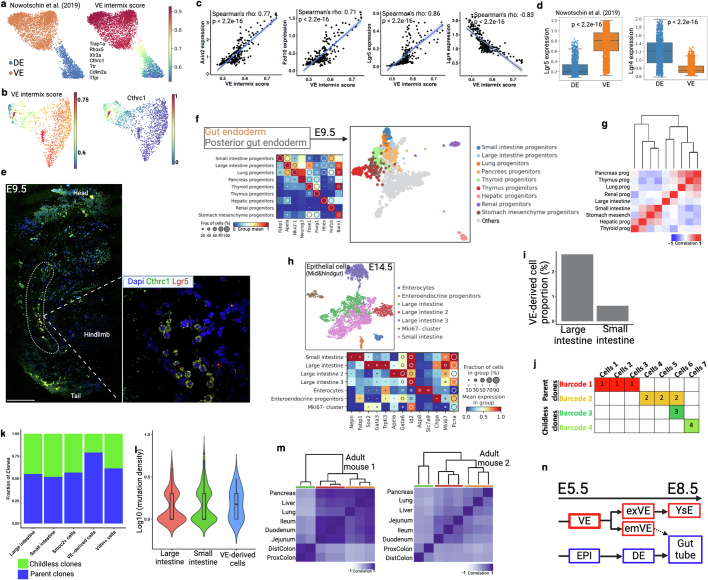
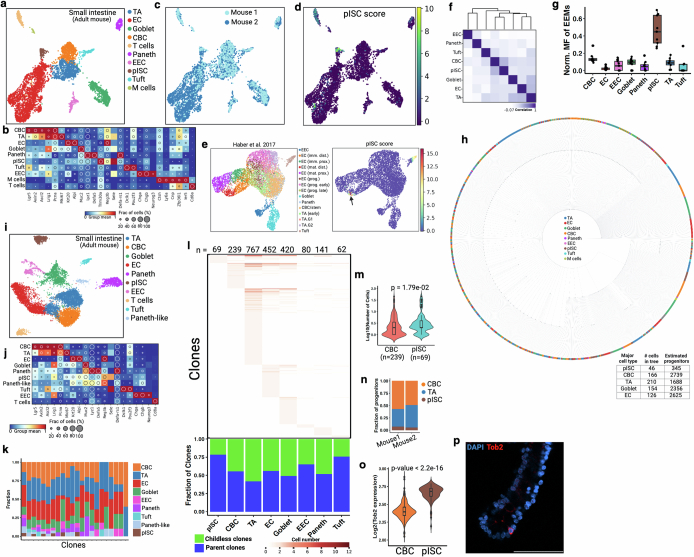
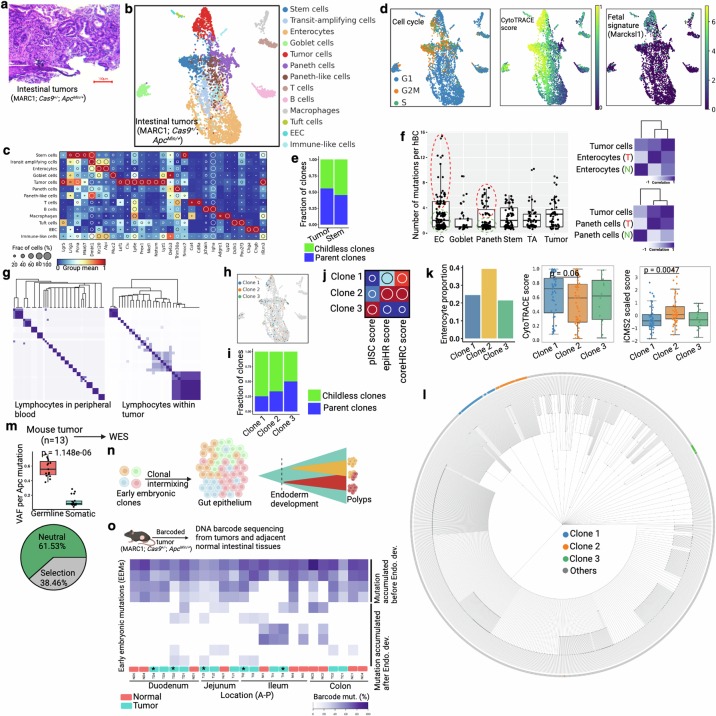
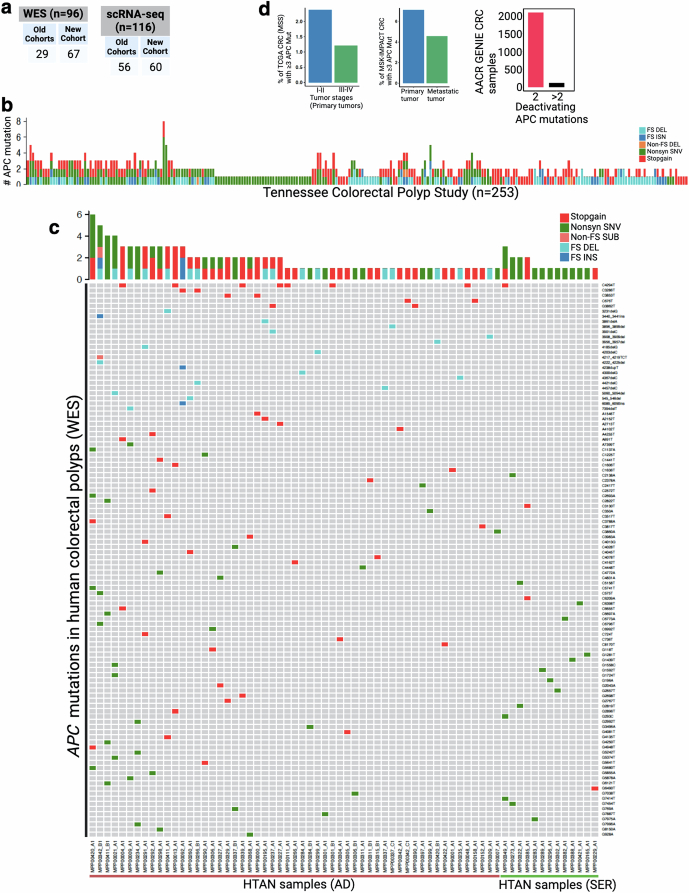
Temporal ordering of cellular events offers fundamental insights into biological phenomena. Although this is traditionally achieved through continuous direct observations1,2, an alternative solution leverages irreversible genetic changes, such as naturally occurring mutations, to create indelible marks that enables retrospective temporal ordering3-5. Using a multipurpose, single-cell CRISPR platform, we developed a molecular clock approach to record the timing of cellular events and clonality in vivo, with incorporation of cell state and lineage information. Using this approach, we uncovered precise timing of tissue-specific cell expansion during mouse embryonic development, unconventional developmental relationships between cell types and new epithelial progenitor states by their unique genetic histories. Analysis of mouse adenomas, coupled to multiomic and single-cell profiling of human precancers, with clonal analysis of 418 human polyps, demonstrated the occurrence of polyclonal initiation in 15-30% of colonic precancers, showing their origins from multiple normal founders. Our study presents a multimodal framework that lays the foundation for in vivo recording, integrating synthetic or natural indelible genetic changes with single-cell analyses, to explore the origins and timing of development and tumorigenesis in mammalian systems.
© 2024. The Author(s).
Conflict of interest statement
M.J.S. received funding from Janssen. J.C.R. is on the scientific advisory board of Sitryx Therapeutics. K.S.L. is an hourly consultant for Etiome, Inc. G.M.C. is a founder of Colossal Biosciences Inc., Dallas, TX. L.T.T. is currently an employee of Genentech. The other authors declare no competing interests.
Figures

Update of
-
Temporal recording of mammalian development and precancer.bioRxiv [Preprint]. 2023 Dec 19:2023.12.18.572260. doi: 10.1101/2023.12.18.572260. bioRxiv. 2023. Update in: Nature. 2024 Oct;634(8036):1187-1195. doi: 10.1038/s41586-024-07954-4. PMID: 38187699 Free PMC article. Updated. Preprint.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
