

Clonal dynamics after allogeneic haematopoietic cell transplantation
- PMID: 39478227
- PMCID: PMC11602715
- DOI: 10.1038/s41586-024-08128-y
Clonal dynamics after allogeneic haematopoietic cell transplantation
Abstract
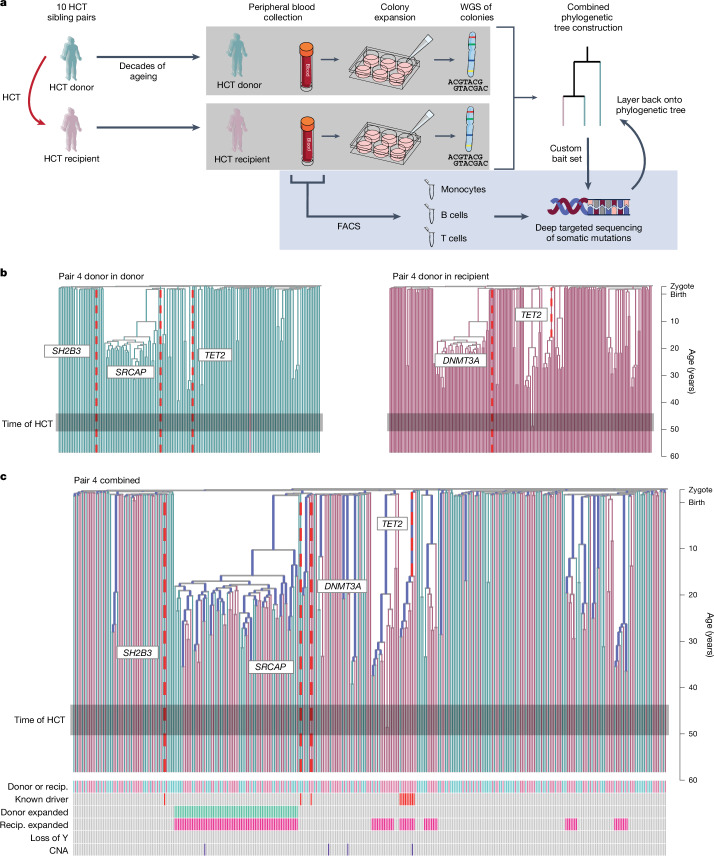
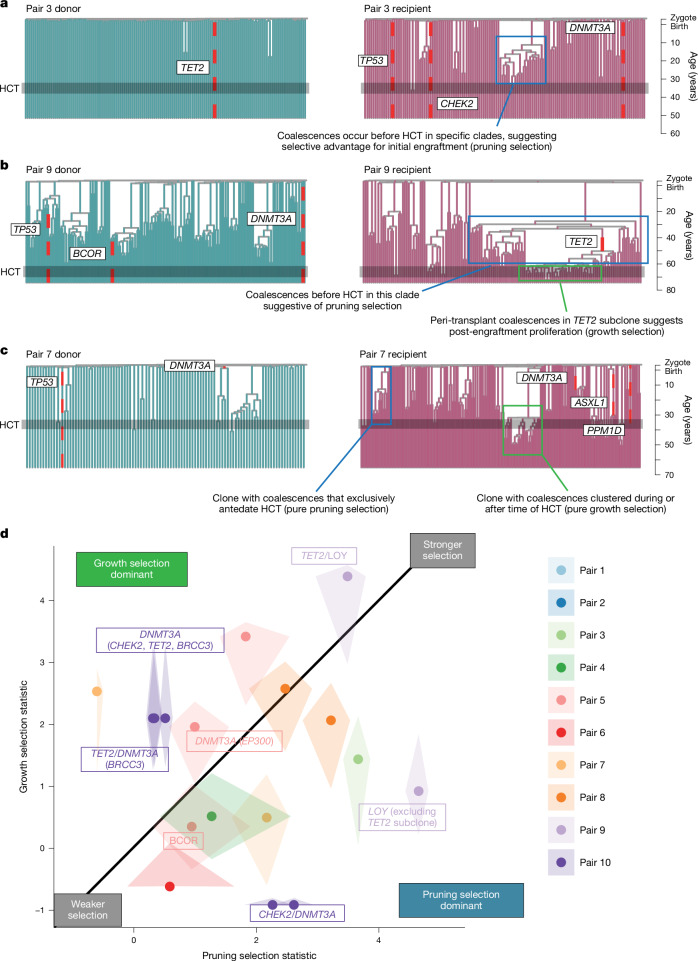
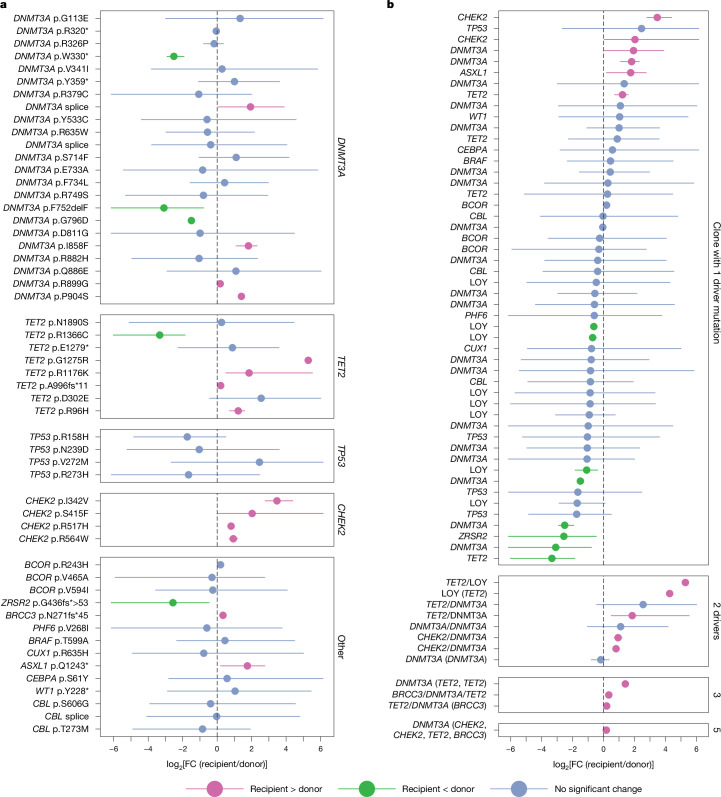
Allogeneic haematopoietic cell transplantation (HCT) replaces the stem cells responsible for blood production with those from a donor1,2. Here, to quantify dynamics of long-term stem cell engraftment, we sequenced genomes from 2,824 single-cell-derived haematopoietic colonies of ten donor-recipient pairs taken 9-31 years after HLA-matched sibling HCT3. With younger donors (18-47 years at transplant), 5,000-30,000 stem cells had engrafted and were still contributing to haematopoiesis at the time of sampling; estimates were tenfold lower with older donors (50-66 years). Engrafted cells made multilineage contributions to myeloid, B lymphoid and T lymphoid populations, although individual clones often showed biases towards one or other mature cell type. Recipients had lower clonal diversity than matched donors, equivalent to around 10-15 years of additional ageing, arising from up to 25-fold greater expansion of stem cell clones. A transplant-related population bottleneck could not explain these differences; instead, phylogenetic trees evinced two distinct modes of HCT-specific selection. In pruning selection, cell divisions underpinning recipient-enriched clonal expansions had occurred in the donor, preceding transplant-their selective advantage derived from preferential mobilization, collection, survival ex vivo or initial homing. In growth selection, cell divisions underpinning clonal expansion occurred in the recipient's marrow after engraftment, most pronounced in clones with multiple driver mutations. Uprooting stem cells from their native environment and transplanting them to foreign soil exaggerates selective pressures, distorting and accelerating the loss of clonal diversity compared to the unperturbed haematopoiesis of donors.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: P.J.C. is a co-founder, stock holder and consultant for Quotient Therapeutics.
Figures













References
-
- Sevilla, J., Fernández-Plaza, S., Diaz, M. A. & Madero, L. Hematopoietic transplantation for bone marrow failure syndromes and thalassemia. Bone Marrow Transplant35, S17–S21 (2005). - PubMed
-
- Boettcher, S. et al. Clonal hematopoiesis in donors and long-term survivors of related allogeneic hematopoietic stem cell transplantation. Blood135, 1548–1559 (2020). - PubMed
-
- Atsuta, Y. et al. Late mortality and causes of death among long-term survivors after allogeneic stem cell transplantation. Biol. Blood Marrow Transplant22, 1702–1709 (2016). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
