

Tumour-intrinsic PDL1 signals regulate the Chk2 DNA damage response in cancer cells and mediate resistance to Chk1 inhibitors
- PMID: 39478560
- PMCID: PMC11523829
- DOI: 10.1186/s12943-024-02147-z
Tumour-intrinsic PDL1 signals regulate the Chk2 DNA damage response in cancer cells and mediate resistance to Chk1 inhibitors
Abstract
Background: Aside from the canonical role of PDL1 as a tumour surface-expressed immune checkpoint molecule, tumour-intrinsic PDL1 signals regulate non-canonical immunopathological pathways mediating treatment resistance whose significance, mechanisms, and therapeutic targeting remain incompletely understood. Recent reports implicate tumour-intrinsic PDL1 signals in the DNA damage response (DDR), including promoting homologous recombination DNA damage repair and mRNA stability of DDR proteins, but many mechanistic details remain undefined.
Methods: We genetically depleted PDL1 from transplantable mouse and human cancer cell lines to understand consequences of tumour-intrinsic PDL1 signals in the DNA damage response. We complemented this work with studies of primary human tumours and inducible mouse tumours. We developed novel approaches to show tumour-intrinsic PDL1 signals in specific subcellular locations. We pharmacologically depleted tumour PDL1 in vivo in mouse models with repurposed FDA-approved drugs for proof-of-concept clinical translation studies.
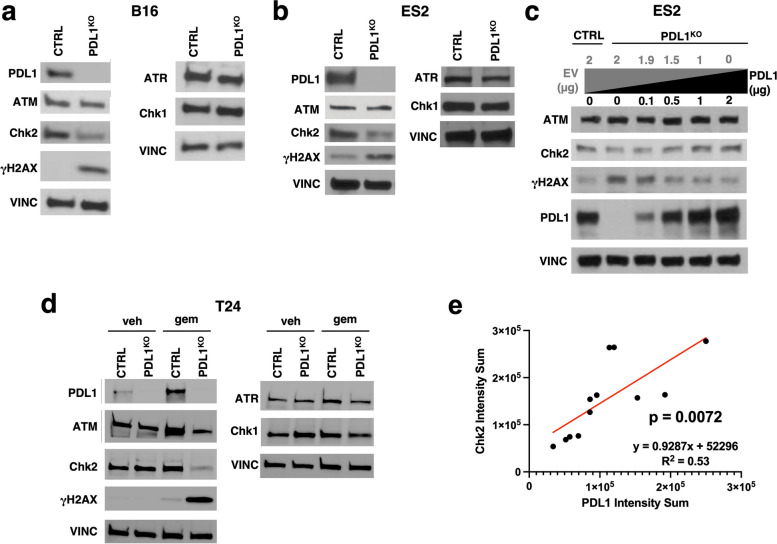
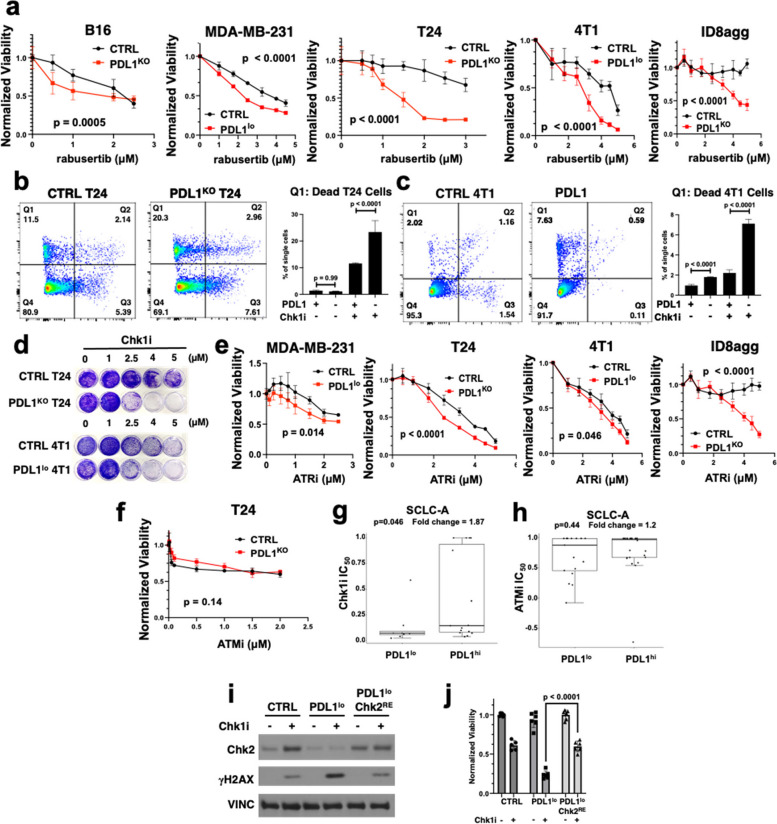
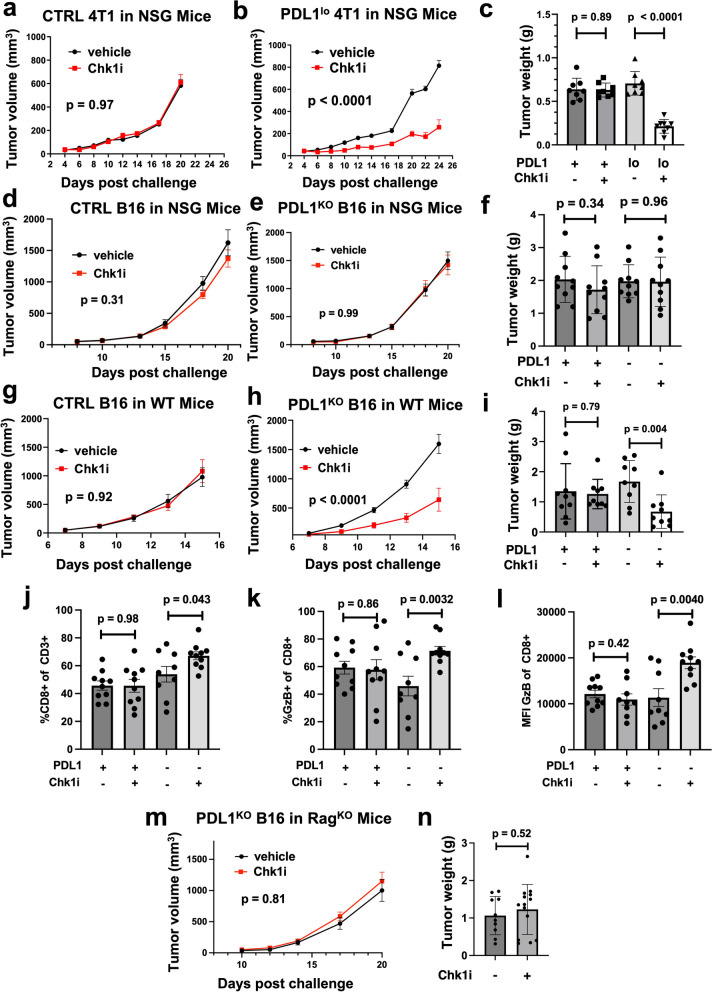
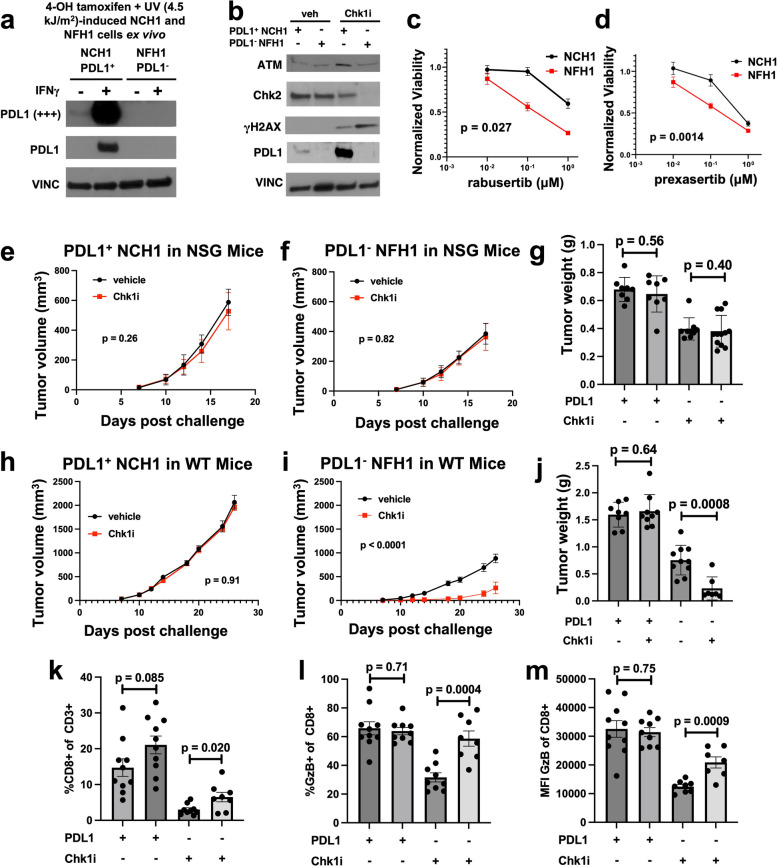
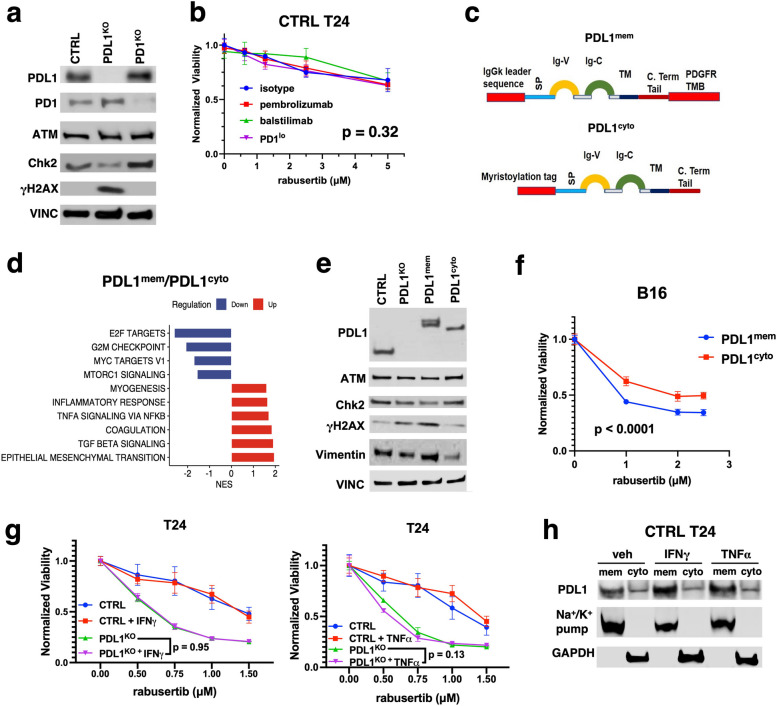
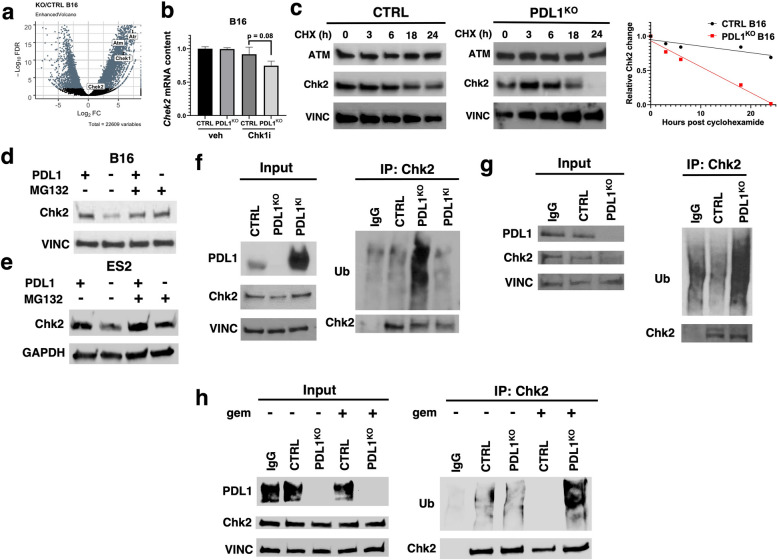
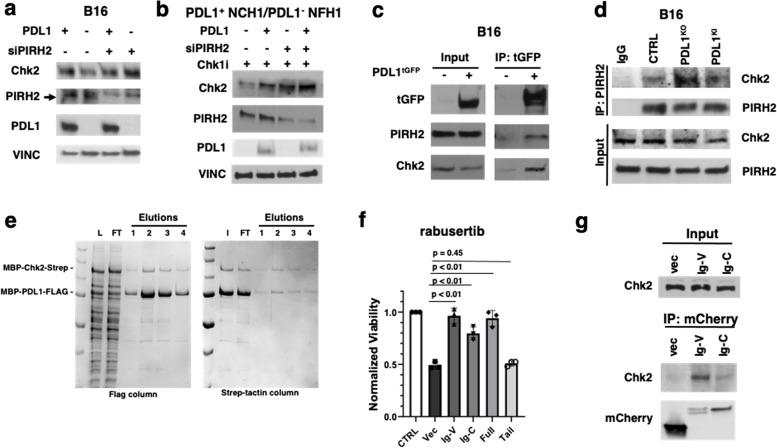
Results: We show that tumour-intrinsic PDL1 promotes the checkpoint kinase-2 (Chk2)-mediated DNA damage response. Intracellular but not surface-expressed PDL1 controlled Chk2 protein content post-translationally and independently of PD1 by antagonising PIRH2 E3 ligase-mediated Chk2 polyubiquitination and protein degradation. Genetic tumour PDL1 depletion specifically reduced tumour Chk2 content but not ATM, ATR, or Chk1 DDR proteins, enhanced Chk1 inhibitor (Chk1i) synthetic lethality in vitro in diverse human and murine tumour models, and improved Chk1i efficacy in vivo. Pharmacologic tumour PDL1 depletion with cefepime or ceftazidime replicated genetic tumour PDL1 depletion by reducing tumour Chk2, inducing Chk1i synthetic lethality in a tumour PDL1-dependent manner, and reducing in vivo tumour growth when combined with Chk1i.
Conclusions: Our data challenge the prevailing surface PDL1 paradigm, elucidate important and previously unappreciated roles for tumour-intrinsic PDL1 in regulating the ATM/Chk2 DNA damage response axis and E3 ligase-mediated protein degradation, suggest tumour PDL1 as a biomarker for Chk1i efficacy, and support the rapid clinical potential of pharmacologic tumour PDL1 depletion to treat selected cancers.
Keywords: Chk2; DDR inhibitors; DNA damage repair; Immune checkpoints; PDL1; Synthetic lethality.
© 2024. The Author(s).
Conflict of interest statement
TJC and AVK have filed a patent on using PDL1 depleting drugs to treat cancer.
Figures

References
-
- Taube JM, et al. Colocalisation of inflammatory response with B7–h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra137. 10.1126/scitranslmed.3003689 https://doi.org:4/127/127ra37 . - DOI - PMC - PubMed
-
- Paterson AM, et al. The programmed death-1 ligand 1:b7–1 pathway restrains diabetogenic effector T cells in vivo. J Immunol. 2011;187:1097–105. 10.4049/jimmunol.1003496 https://doi.org:jimmunol.1003496 . - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
