

HCMV strain- and cell type-specific alterations in membrane contact sites point to the convergent regulation of organelle remodeling
- PMID: 39480111
- PMCID: PMC11575408
- DOI: 10.1128/jvi.01099-24
HCMV strain- and cell type-specific alterations in membrane contact sites point to the convergent regulation of organelle remodeling
Abstract
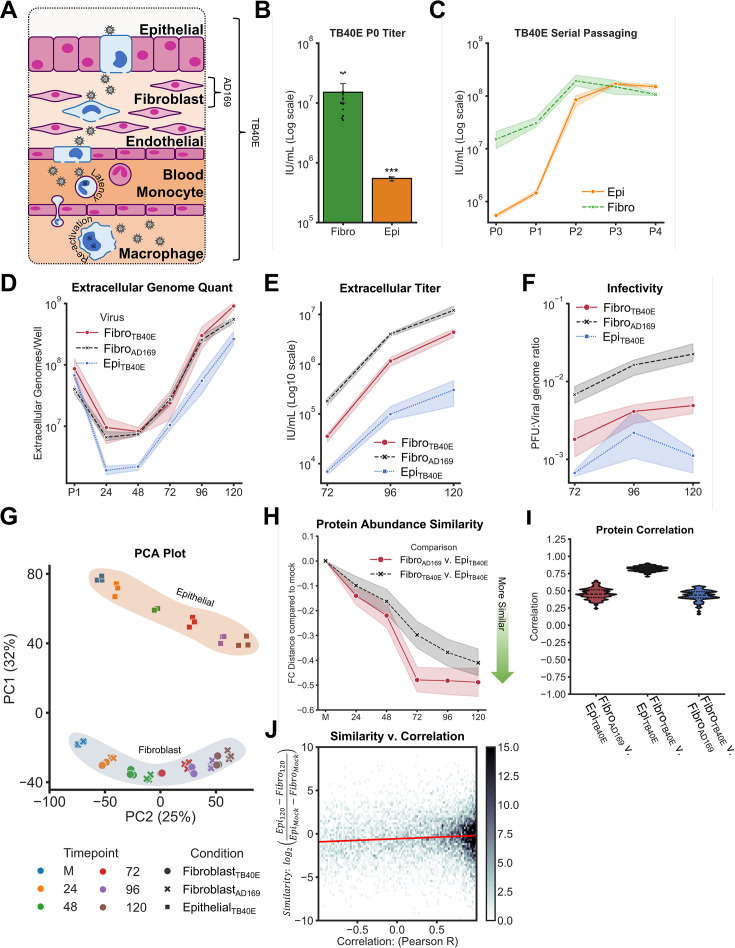
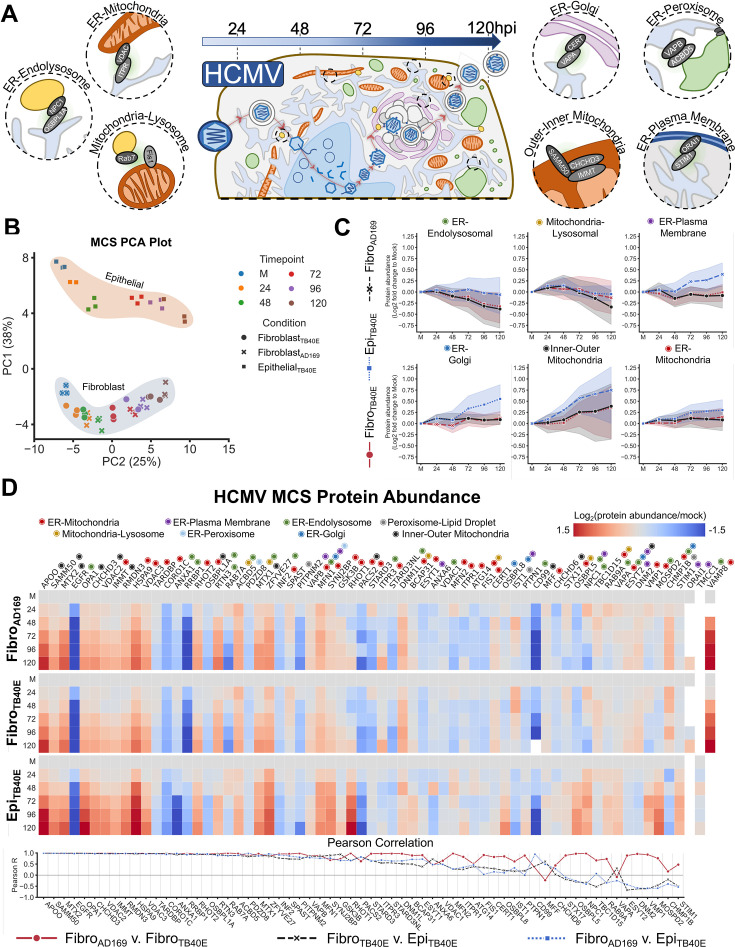
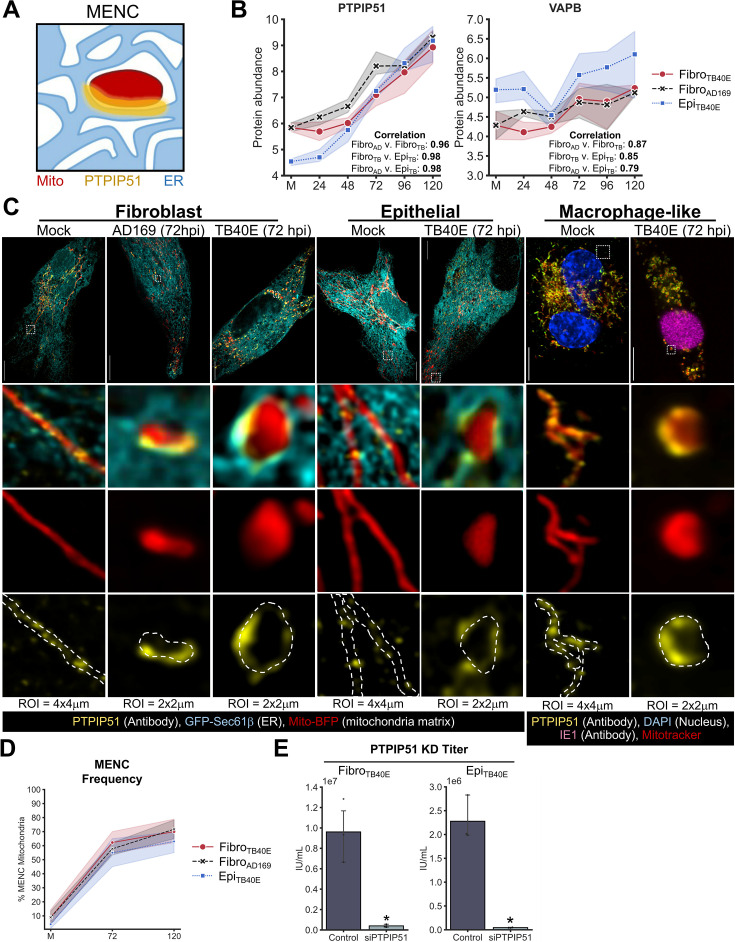
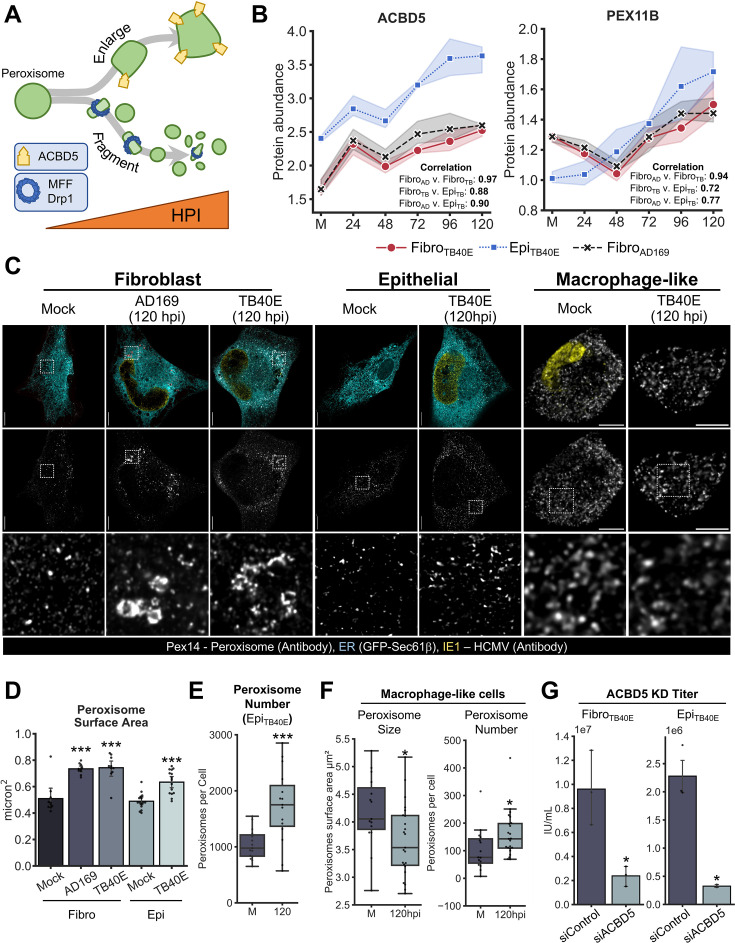
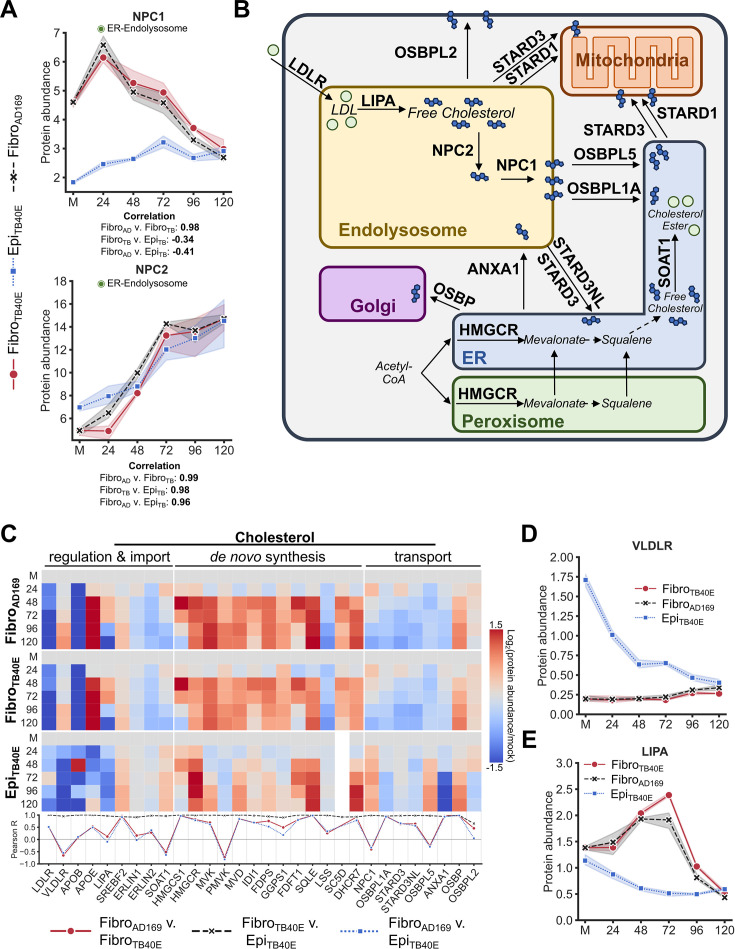
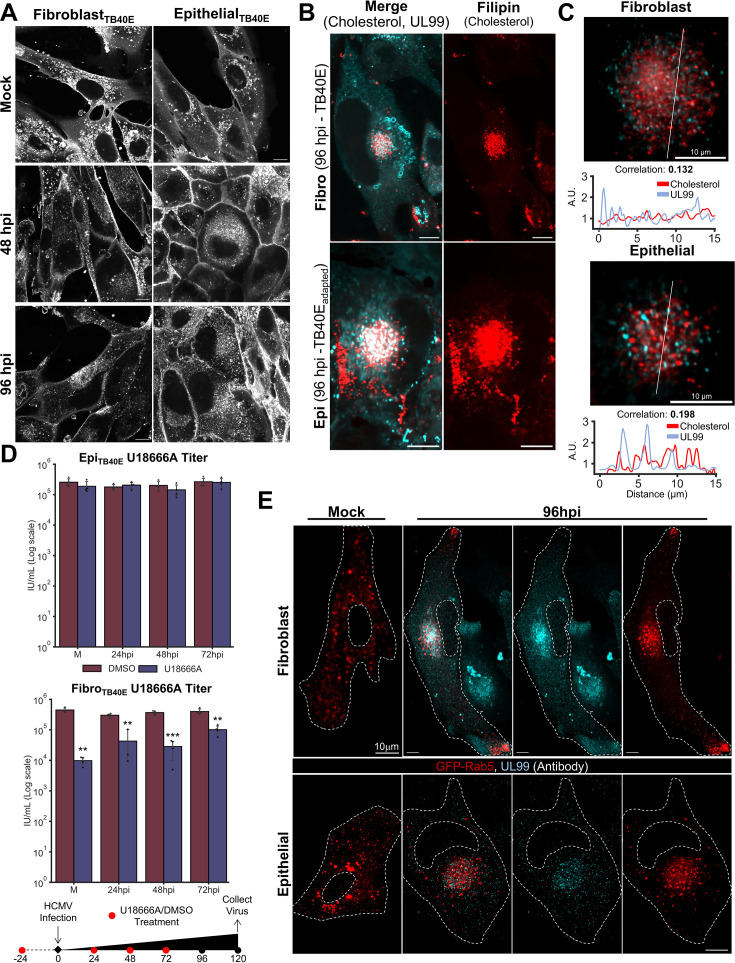
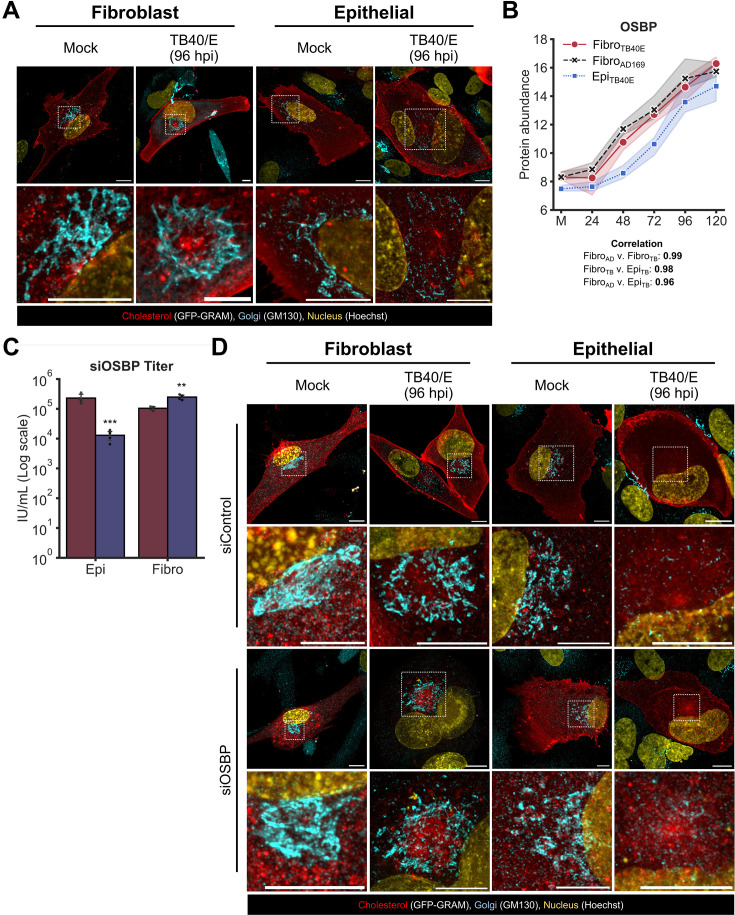
Viruses are ubiquitous entities that infect organisms across the kingdoms of life. While viruses can infect a range of cells, tissues, and organisms, this aspect is often not explored in cell culture analyses. There is limited information about which infection-induced changes are shared or distinct in different cellular environments. The prevalent pathogen human cytomegalovirus (HCMV) remodels the structure and function of subcellular organelles and their interconnected networks formed by membrane contact sites (MCSs). A large portion of this knowledge has been derived from fibroblasts infected with a lab-adapted HCMV strain. Here, we assess strain- and cell type-specific alterations in MCSs and organelle remodeling induced by HCMV. Integrating quantitative mass spectrometry, super-resolution microscopy, and molecular virology assays, we compare infections with lab-adapted and low-passage HCMV strains in fibroblast and epithelial cells. We determine that, despite baseline proteome disparities between uninfected fibroblast and epithelial cells, infection induces convergent changes and is remarkably similar. We show that hallmarks of HCMV infection in fibroblasts, mitochondria-endoplasmic reticulum (ER) encapsulations and peroxisome proliferation, are also conserved in infected epithelial and macrophage-like cells. Exploring cell type-specific differences, we demonstrate that fibroblasts rely on endosomal cholesterol transport while epithelial cells rely on cholesterol from the Golgi. Despite these mechanistic differences, infections in both cell types result in phenotypically similar cholesterol accumulation at the viral assembly complex. Our findings highlight the adaptability of HCMV, in that infections can be tailored to the initial cell state by inducing both shared and unique proteome alterations, ultimately promoting a unified pro-viral environment.IMPORTANCEHuman cytomegalovirus (HCMV) establishes infections in diverse cell types throughout the body and is connected to a litany of diseases associated with each of these tissues. However, it is still not fully understood how HCMV replication varies in distinct cell types. Here, we compare HCMV replication with lab-adapted and low-passage strains in two primary sites of infection, lung fibroblasts and retinal epithelial cells. We discover that, despite displaying disparate protein compositions prior to infection, these cell types undergo convergent alterations upon HCMV infection, reaching a more similar cellular state late in infection. We find that remodeling of the subcellular landscape is a pervasive feature of HCMV infection, through alterations to both organelle structure-function and the interconnected networks they form via membrane contact sites. Our findings show how HCMV infection in different cell types induces both shared and divergent changes to cellular processes, ultimately leading to a more unified state.
Keywords: HCMV; herpesvirus; membrane contact sites; organelle remodeling; proteomics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Pila-Castellanos I, Molino D, McKellar J, Lines L, Da Graca J, Tauziet M, Chanteloup L, Mikaelian I, Meyniel-Schicklin L, Codogno P, Vonderscher J, Delevoye C, Moncorgé O, Meldrum E, Goujon C, Morel E, de Chassey B. 2021. Correction: Mitochondrial morphodynamics alteration induced by influenza virus infection as a new antiviral strategy. PLoS Pathog 17:e1009485. doi: 10.1371/journal.ppat.1009485 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01GM114141/HHS | NIH | National Institute of General Medical Sciences (NIGMS)
- T32GM007388/HHS | National Institutes of Health (NIH)
- TL1TR003019/HHS | NIH | National Center for Advancing Translational Sciences (NCATS)
- T32 GM007388/GM/NIGMS NIH HHS/United States
- 23PRE1014367/American Heart Association (AHA)
- 3.1416/EIF | Stand Up To Cancer (SU2C)
- 2021316158/National Science Foundation (NSF)
- R01 AI174515/AI/NIAID NIH HHS/United States
- R01 GM114141/GM/NIGMS NIH HHS/United States
- Distinguished Investigator/Paul G. Allen Frontiers Group (Allen Institute)
- R01AI174515/HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID)
- TL1 TR003019/TR/NCATS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
