

Statistical inference with a manifold-constrained RNA velocity model uncovers cell cycle speed modulations
- PMID: 39482463
- PMCID: PMC11621032
- DOI: 10.1038/s41592-024-02471-8
Statistical inference with a manifold-constrained RNA velocity model uncovers cell cycle speed modulations
Abstract
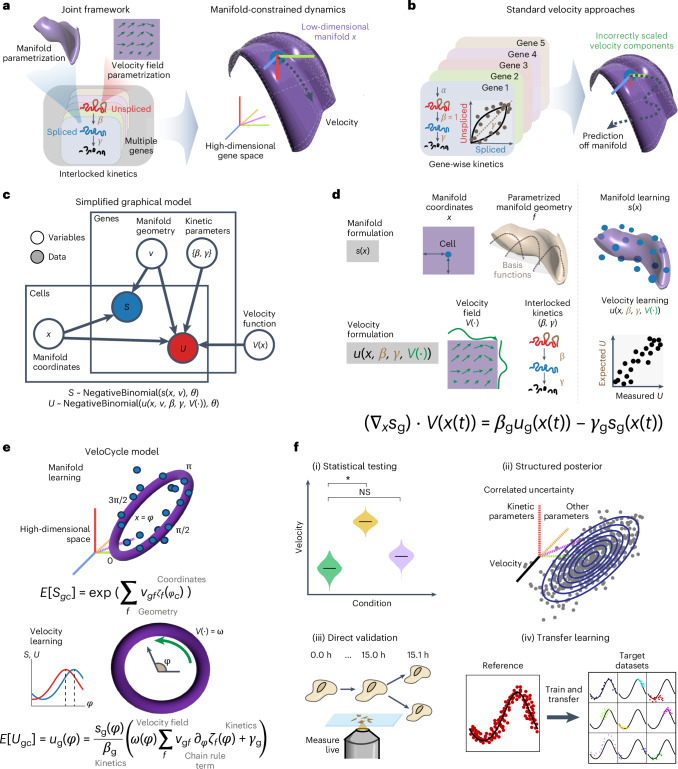
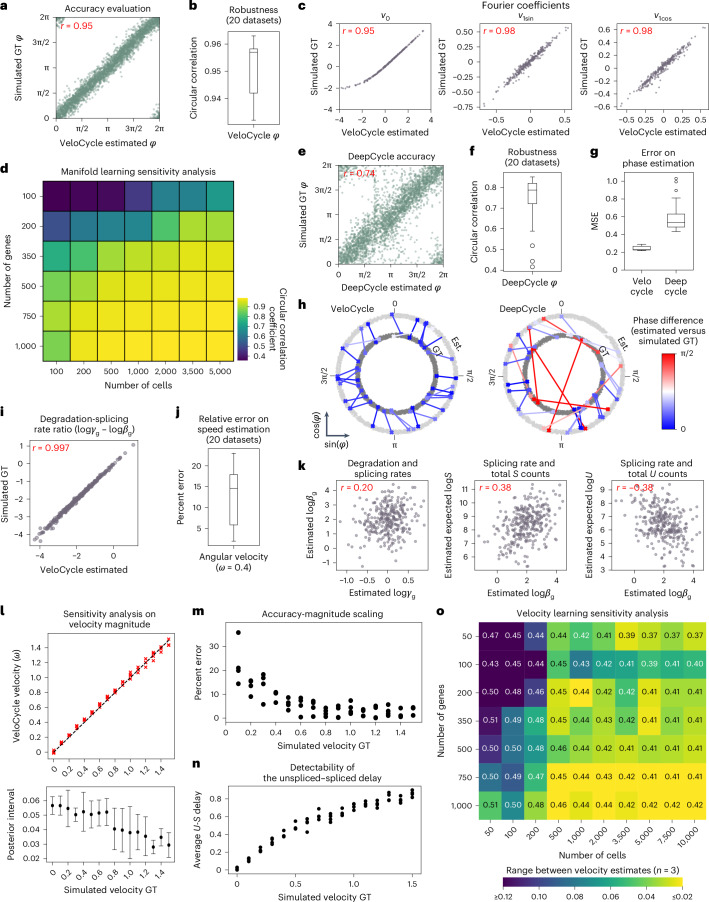
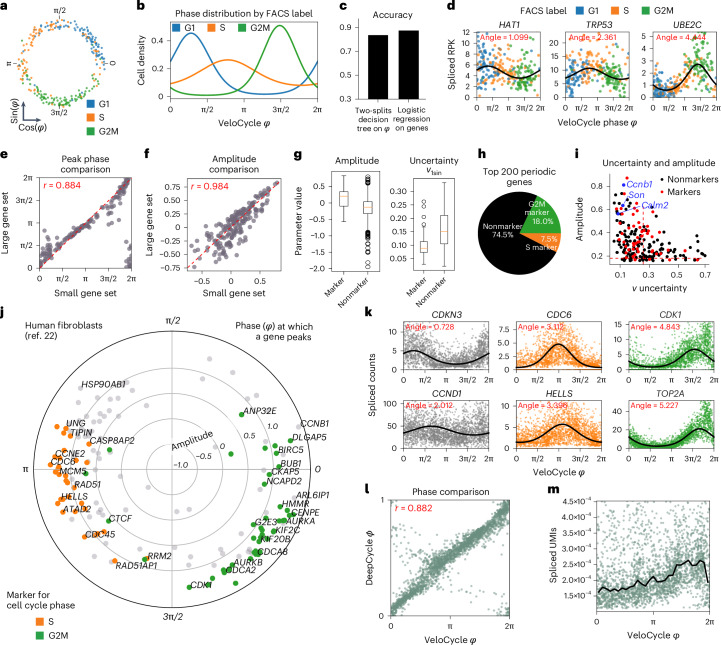
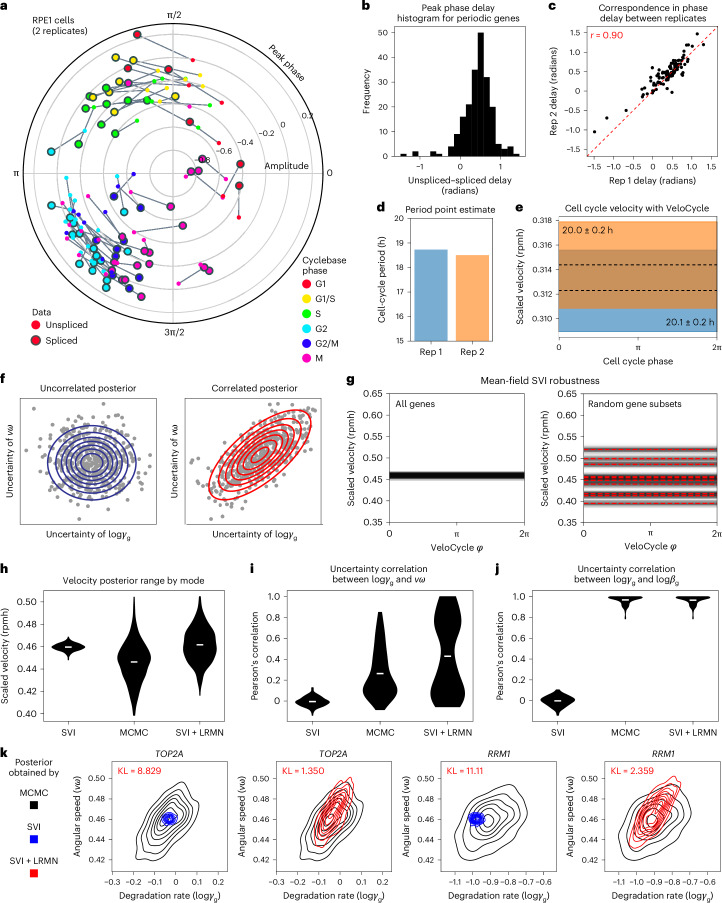
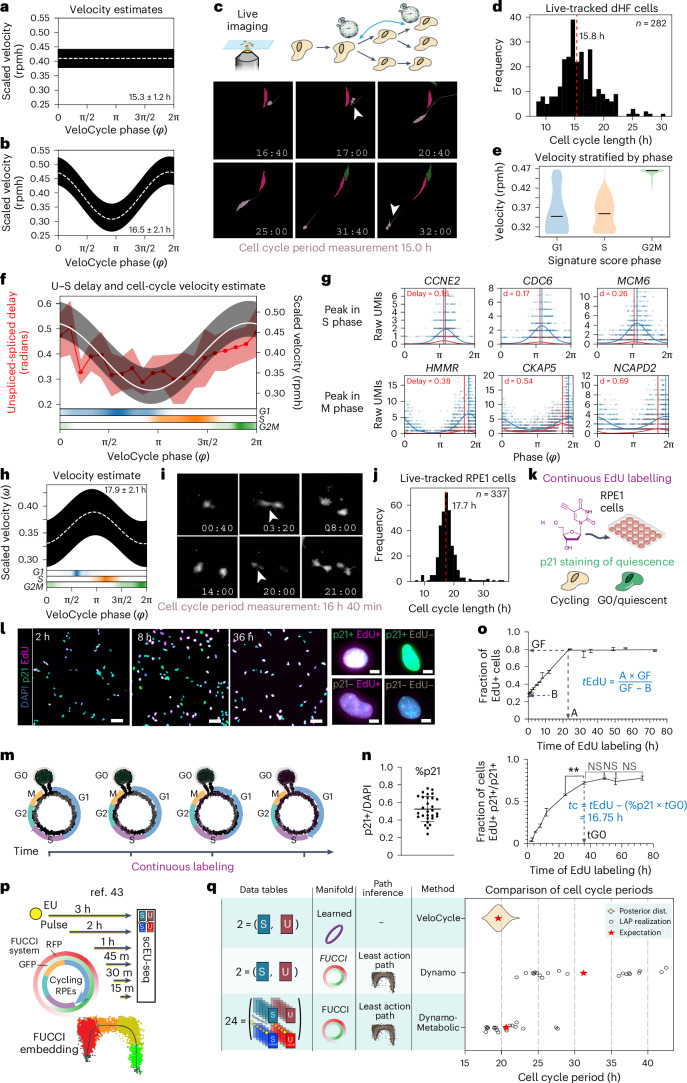
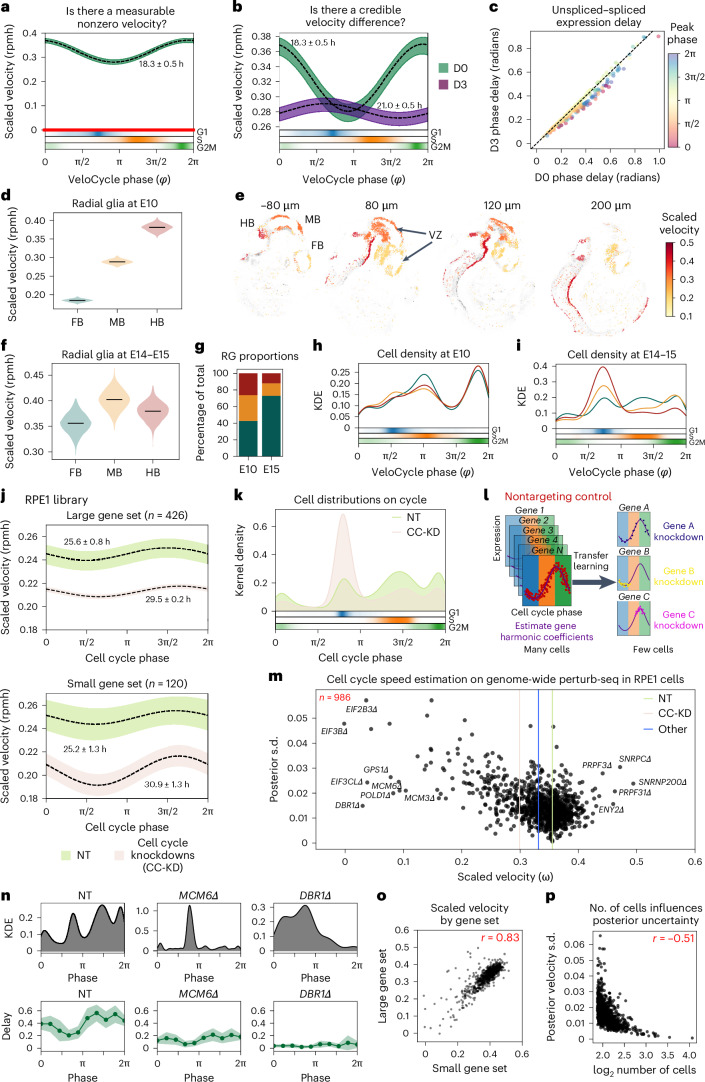
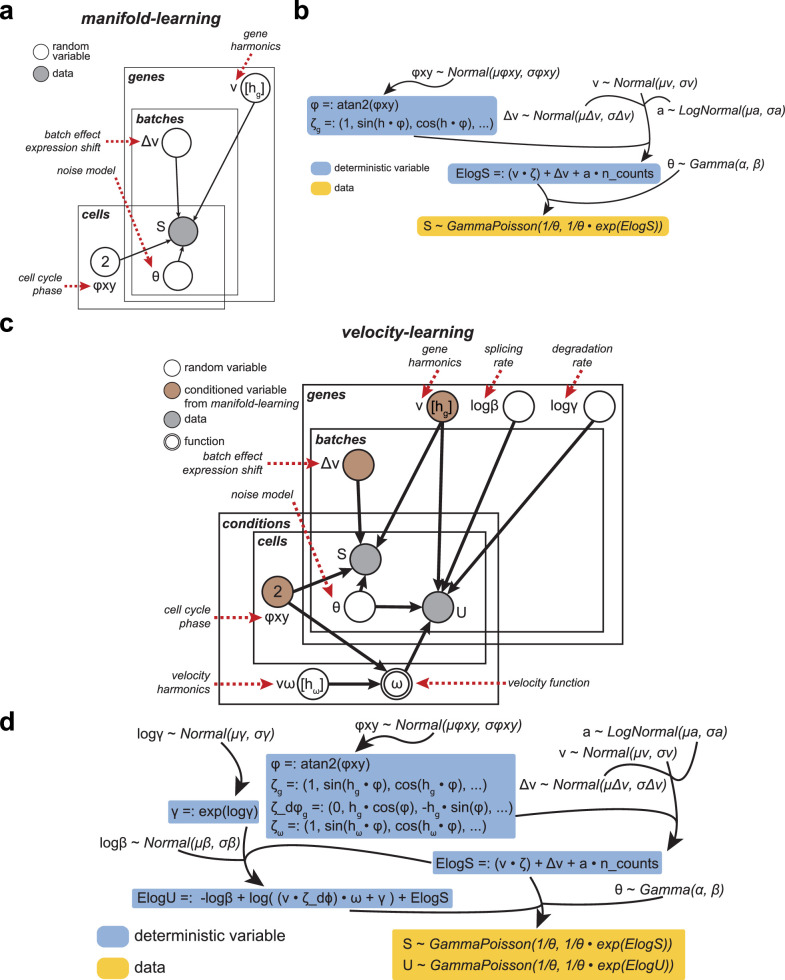
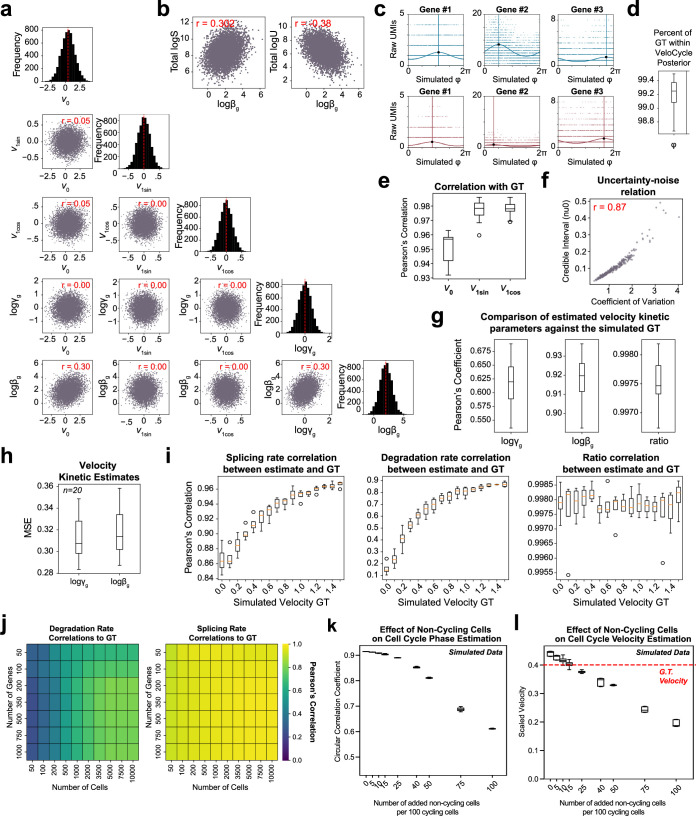
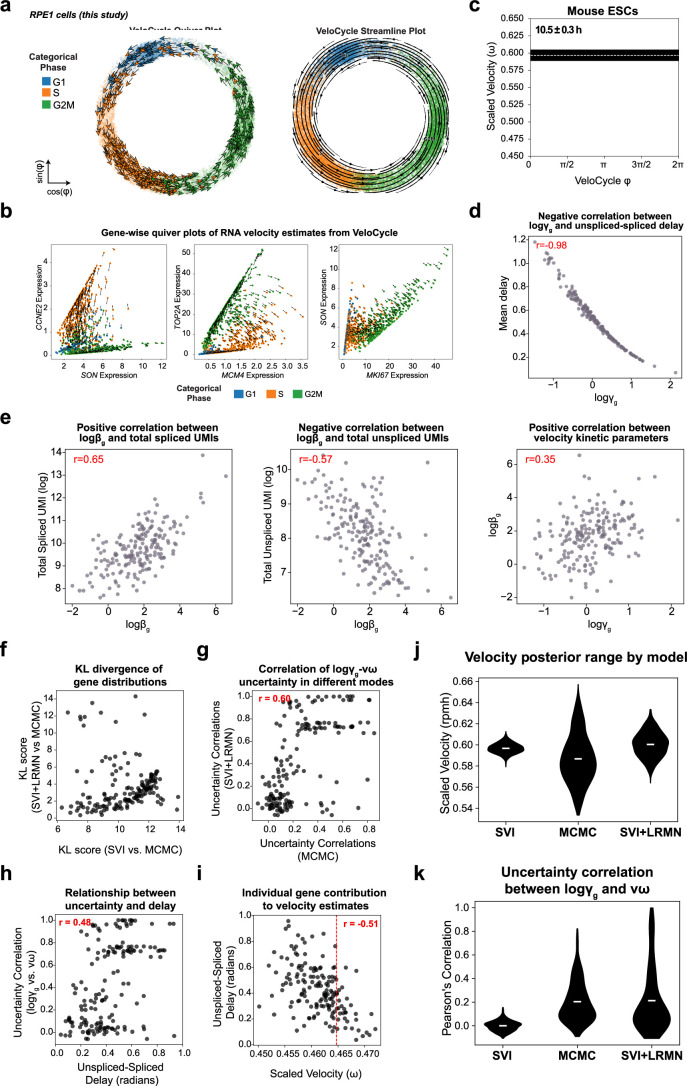
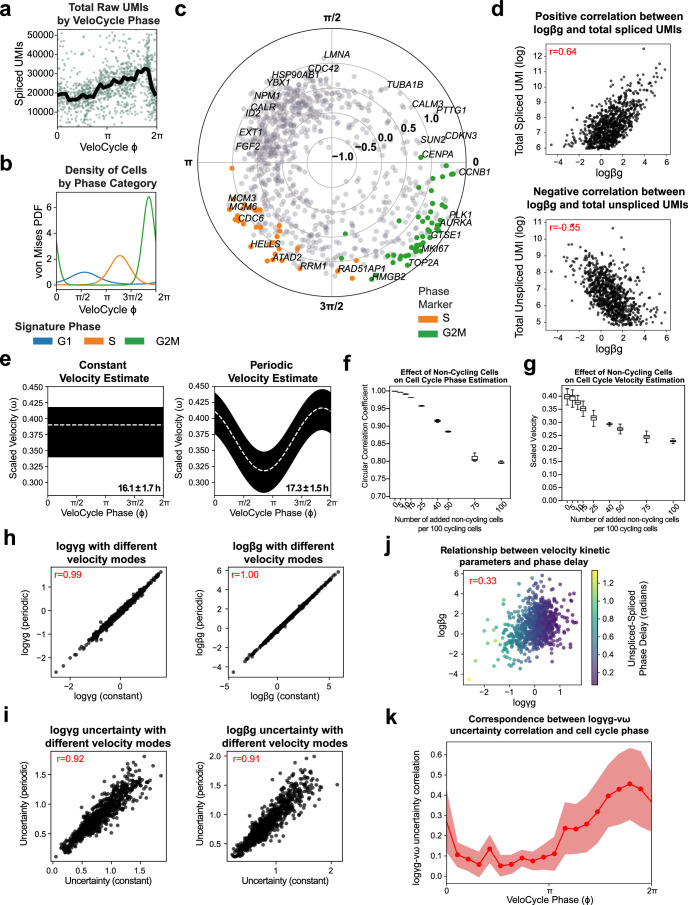
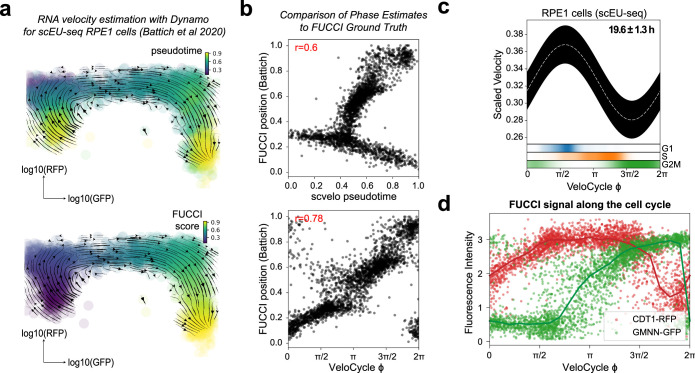
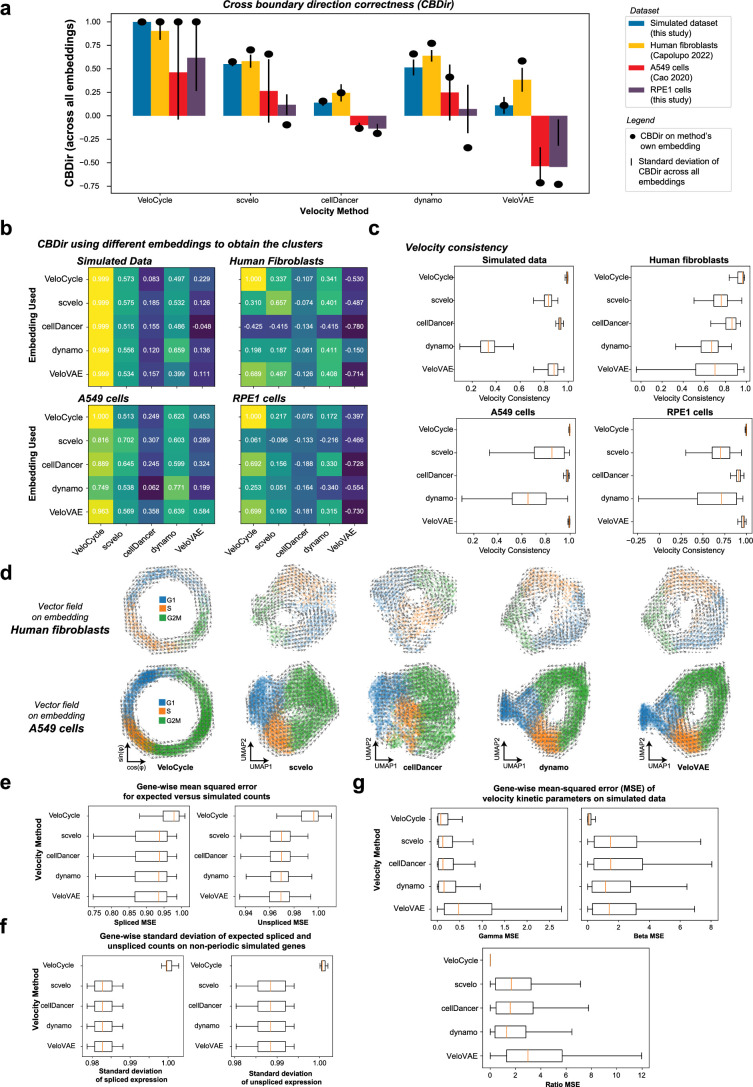
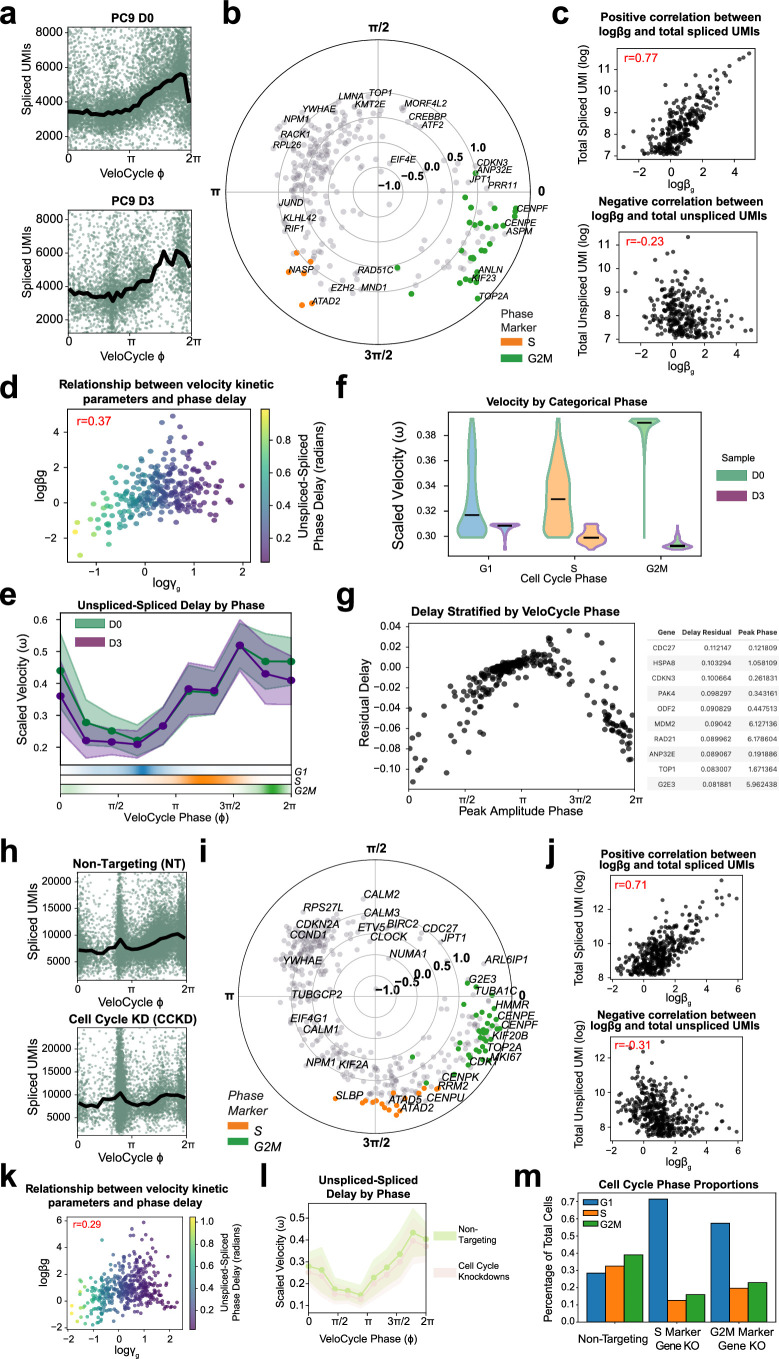
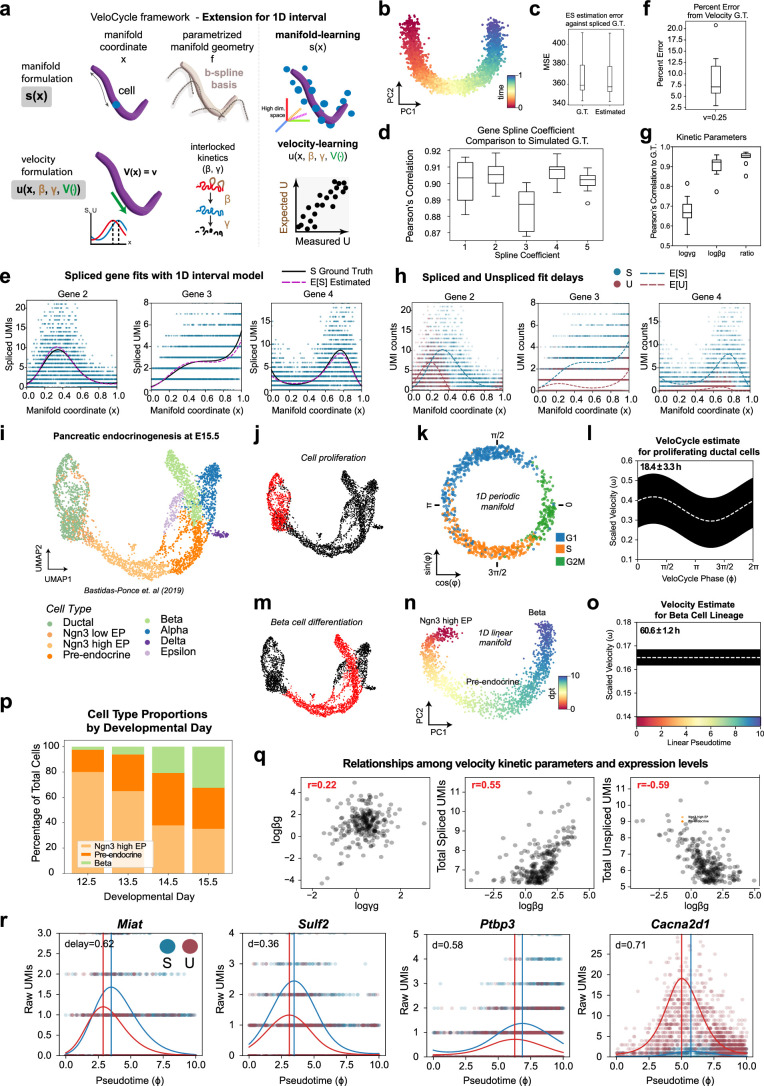
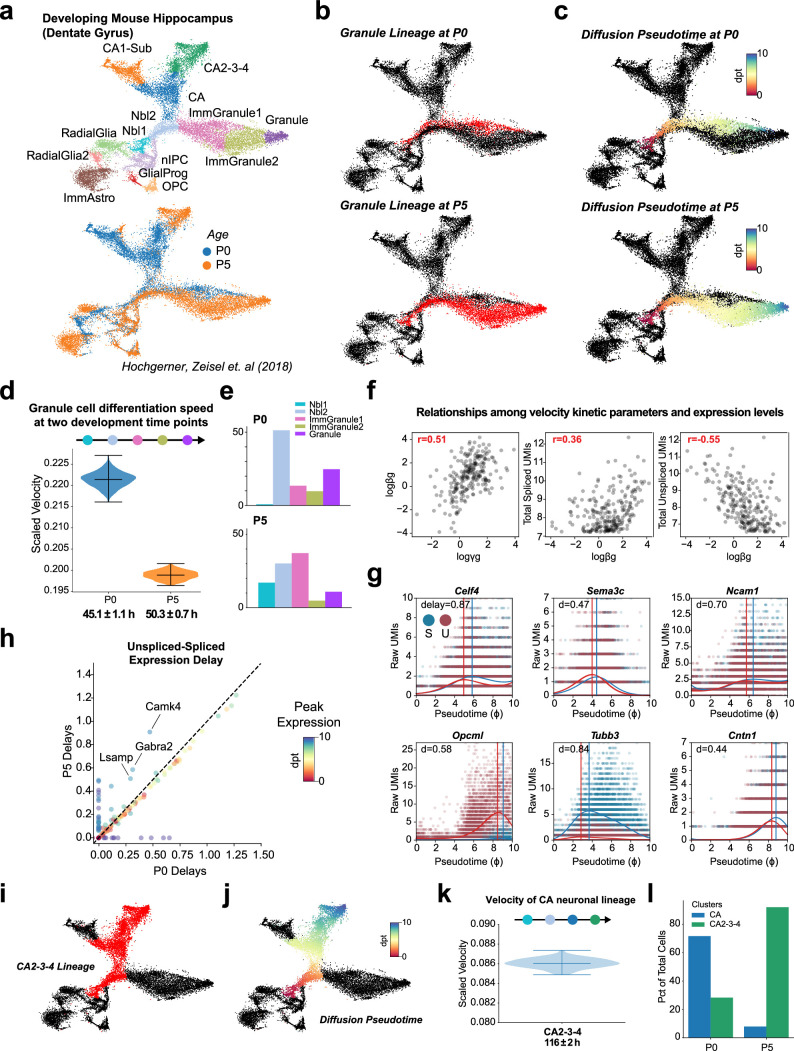
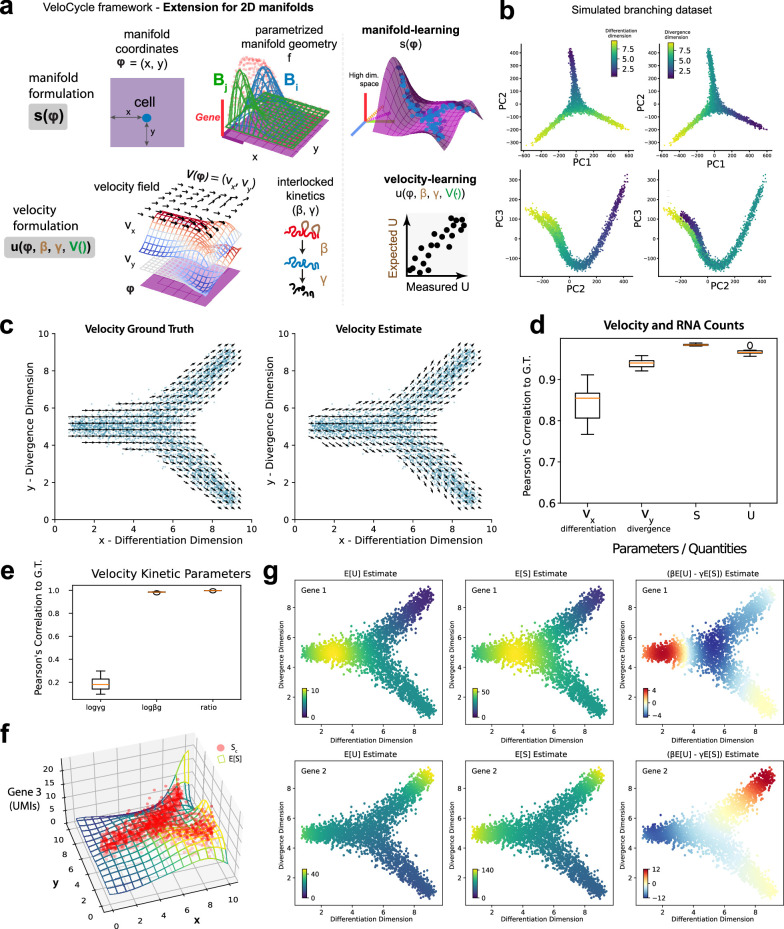
Across biological systems, cells undergo coordinated changes in gene expression, resulting in transcriptome dynamics that unfold within a low-dimensional manifold. While low-dimensional dynamics can be extracted using RNA velocity, these algorithms can be fragile and rely on heuristics lacking statistical control. Moreover, the estimated vector field is not dynamically consistent with the traversed gene expression manifold. To address these challenges, we introduce a Bayesian model of RNA velocity that couples velocity field and manifold estimation in a reformulated, unified framework, identifying the parameters of an explicit dynamical system. Focusing on the cell cycle, we implement VeloCycle to study gene regulation dynamics on one-dimensional periodic manifolds and validate its ability to infer cell cycle periods using live imaging. We also apply VeloCycle to reveal speed differences in regionally defined progenitors and Perturb-seq gene knockdowns. Overall, VeloCycle expands the single-cell RNA sequencing analysis toolkit with a modular and statistically consistent RNA velocity inference framework.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Update of
-
Statistical inference with a manifold-constrained RNA velocity model uncovers cell cycle speed modulations.bioRxiv [Preprint]. 2024 Jan 22:2024.01.18.576093. doi: 10.1101/2024.01.18.576093. bioRxiv. 2024. Update in: Nat Methods. 2024 Dec;21(12):2271-2286. doi: 10.1038/s41592-024-02471-8. PMID: 38328127 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
- R35 HG010717/HG/NHGRI NIH HHS/United States
- PZ00P3_193445/Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (Swiss National Science Foundation)
- 310030B_201267/Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (Swiss National Science Foundation)
- R35HG010717/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
