

MYH1 deficiency disrupts outer hair cell electromotility, resulting in hearing loss
- PMID: 39482536
- PMCID: PMC11612406
- DOI: 10.1038/s12276-024-01338-4
MYH1 deficiency disrupts outer hair cell electromotility, resulting in hearing loss
Abstract
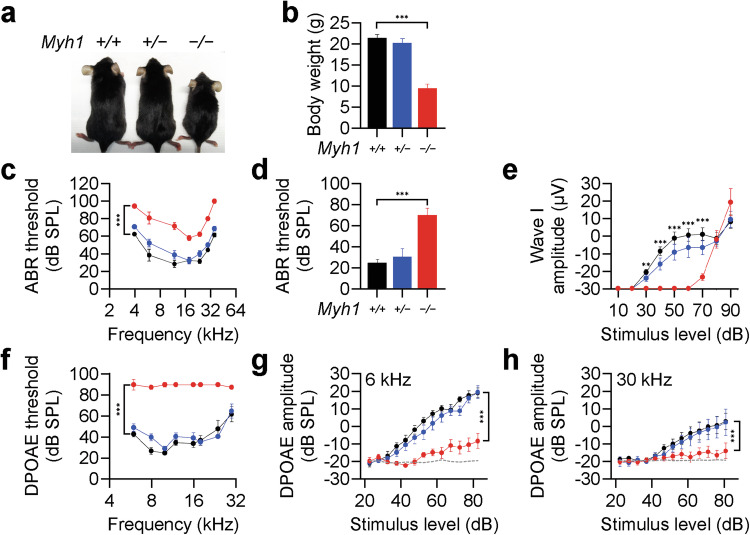
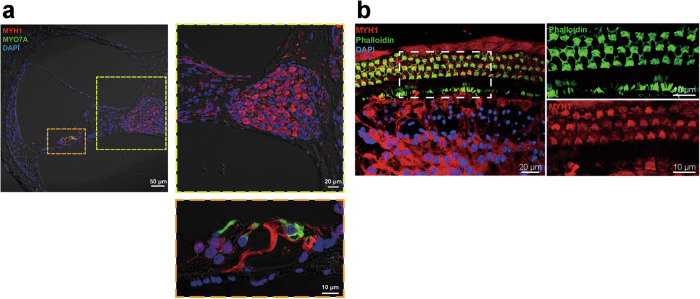
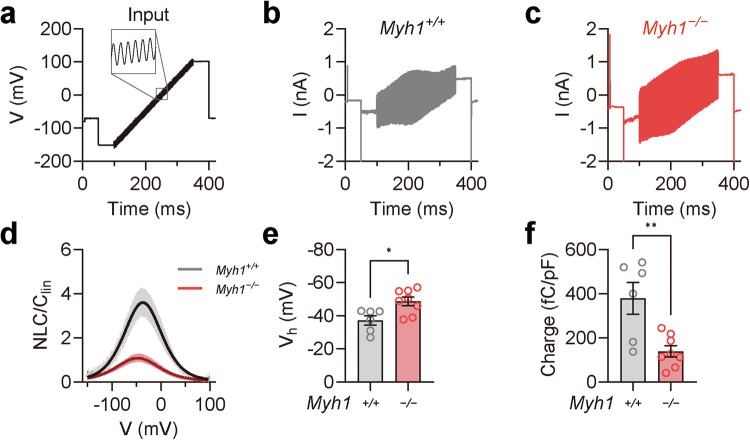
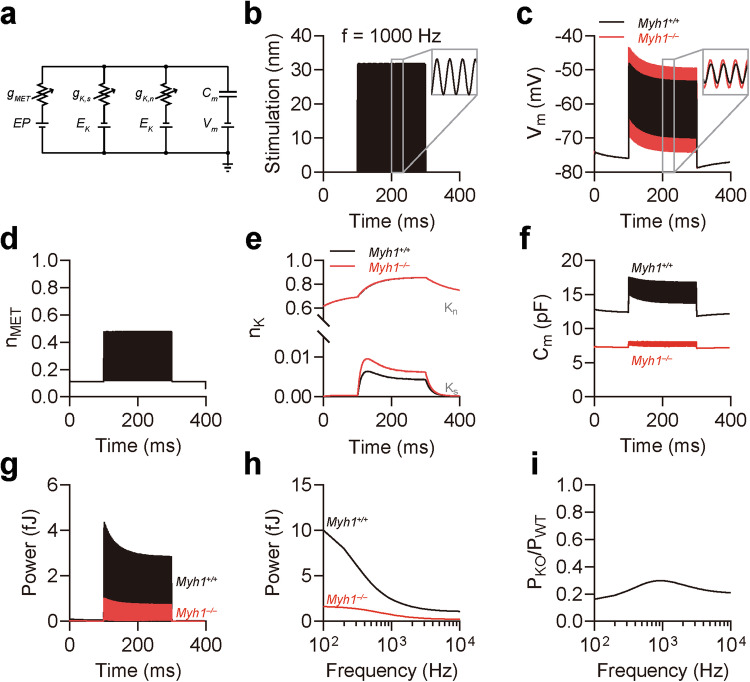
Myh1 is a mouse deafness gene with an unknown function in the auditory system. Hearing loss in Myh1-knockout mice is characterized by an elevated threshold for the auditory brainstem response and the absence of a threshold for distortion product otoacoustic emission. Here, we investigated the role of MYH1 in outer hair cells (OHCs), crucial structures in the organ of Corti responsible for regulating cochlear amplification. Direct whole-cell voltage-clamp recordings of OHCs revealed that prestin activity was lower in Myh1-knockout mice than in wild-type mice, indicating abnormal OHC electromotility. We analyzed whole-exome sequencing data from 437 patients with hearing loss of unknown genetic causes and identified biallelic missense variants of MYH1 in five unrelated families. Hearing loss in individuals harboring biallelic MYH1 variants was non-progressive, with an onset ranging from congenital to childhood. Three of five individuals with MYH1 variants displayed osteopenia. Structural prediction by AlphaFold2 followed by molecular dynamic simulations revealed that the identified variants presented structural abnormalities compared with wild-type MYH1. In a heterogeneous overexpression system, MYH1 variants, particularly those in the head domain, abolished MYH1 functions, such as by increasing prestin activity and modulating the membrane traction force. Overall, our findings suggest an essential function of MYH1 in OHCs, as observed in Myh1-deficient mice, and provide genetic evidence linking biallelic MYH1 variants to autosomal recessive hearing loss in humans.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
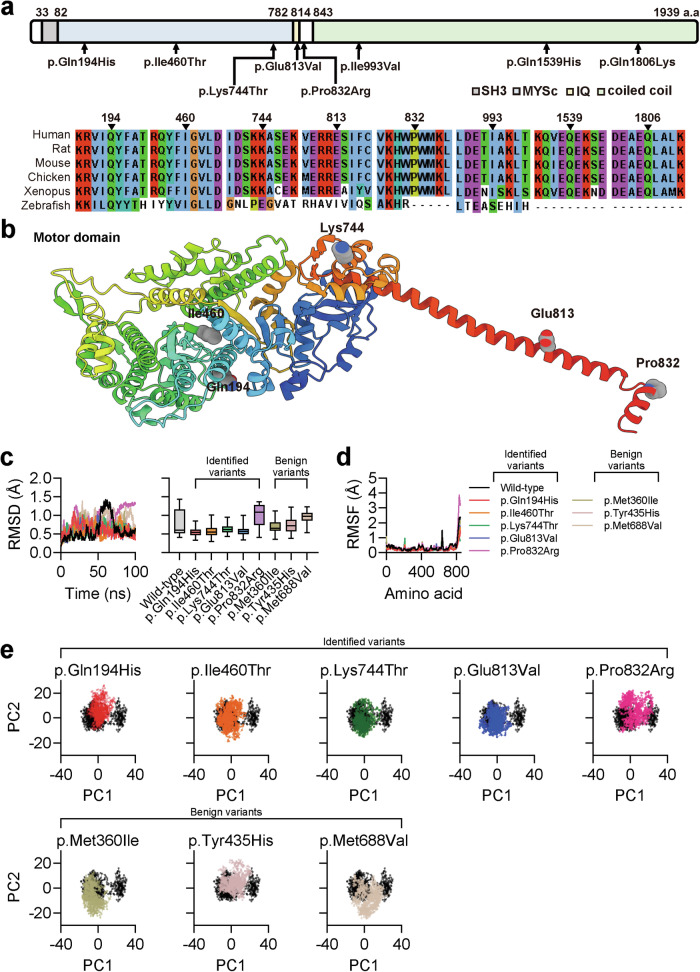
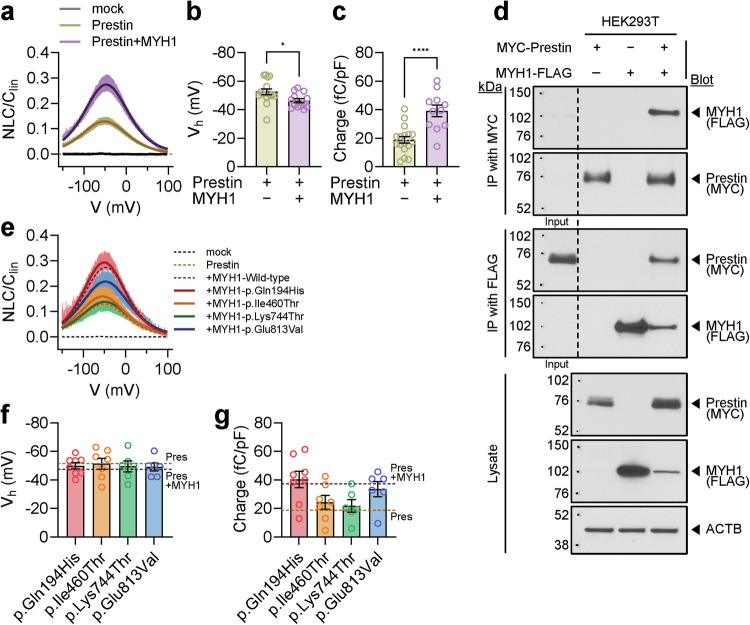
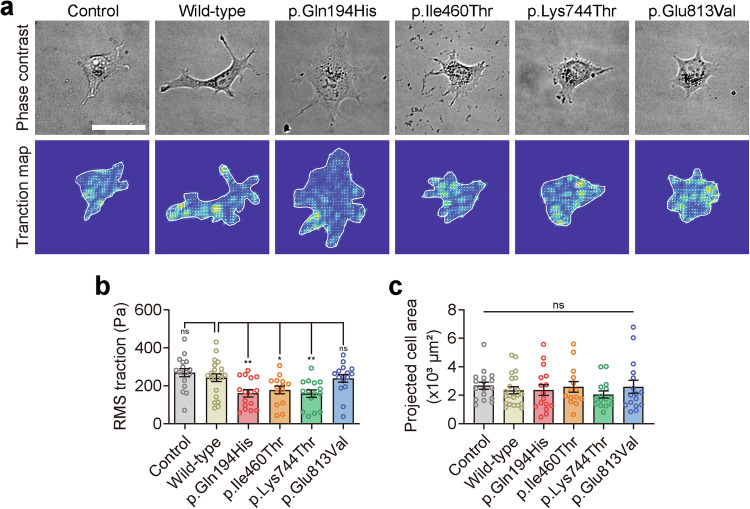
References
-
- Shearer, A. E., Hildebrand, M. S., Schaefer, A. M., & Smith, R. J. H. Genetic Hearing Loss Overview. In M.P. Adam (Eds.) et. al., GeneReviews®. University of Washington, Seattle (1999).
MeSH terms
Substances
Grants and funding
- 2018R1A5A2025079/National Research Foundation of Korea (NRF)
- 2020M3A9D5A01082439/National Research Foundation of Korea (NRF)
- 2020R1A2C3005787/National Research Foundation of Korea (NRF)
- 2022R1A2C3007281/National Research Foundation of Korea (NRF)
- 6-2021-0003/Yonsei University | Yonsei University College of Medicine (YUCM)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
