

Progressive myoclonic ataxia as an initial symptom of typical type I sialidosis with NEU1 mutation
- PMID: 39482827
- PMCID: PMC11572746
- DOI: 10.1002/acn3.52212
Progressive myoclonic ataxia as an initial symptom of typical type I sialidosis with NEU1 mutation
Abstract
Objective: Expand genetic screening for atypical Type I sialidosis (ST-1) could address its underdiagnosed in both progressive myoclonic ataxia (PMA) and ataxia patients. To evaluate the potential founder effect of mutation in the population.
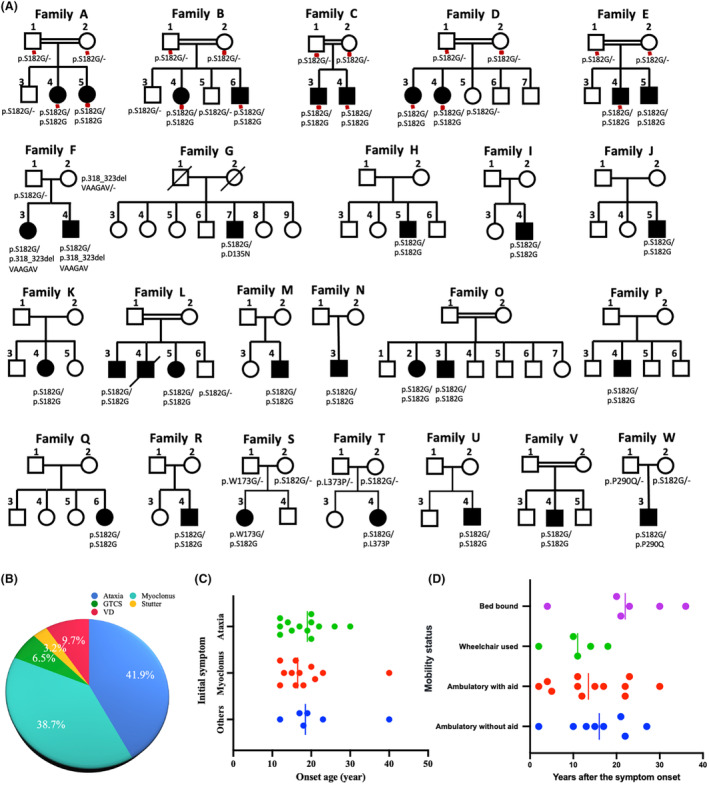
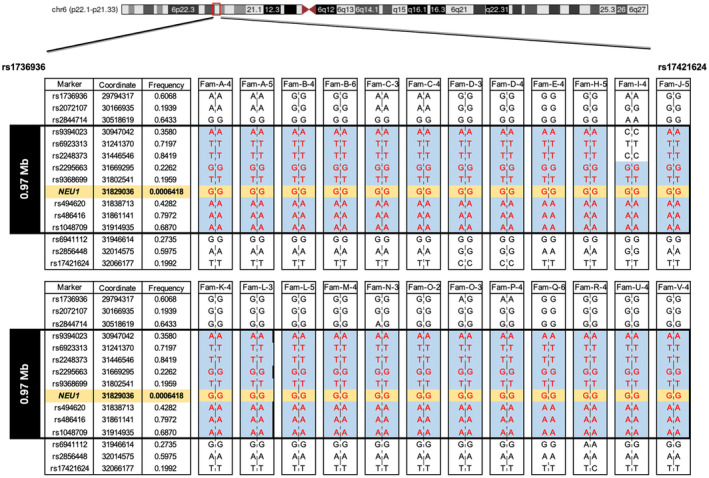
Methods: We enrolled 231 patients with PMA or ataxia from the First Affiliated Hospital of Fujian Medical University. Through Whole Exome Sequencing and Sanger sequencing, we identified the causative gene in patients. Haplotype analysis was employed to explore a potential founder effect of the NEU1 c.544A>G mutation.
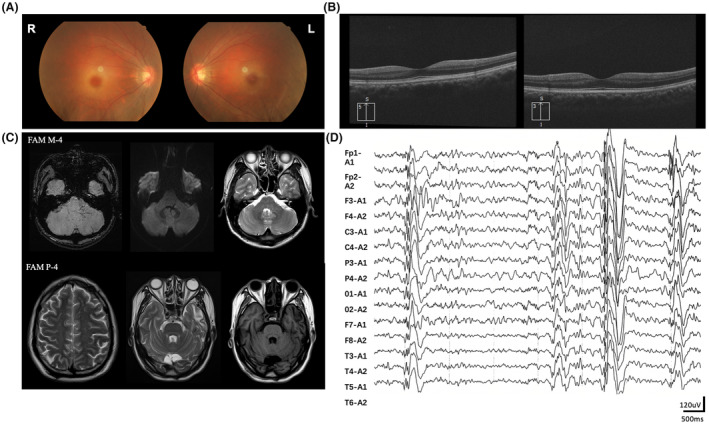
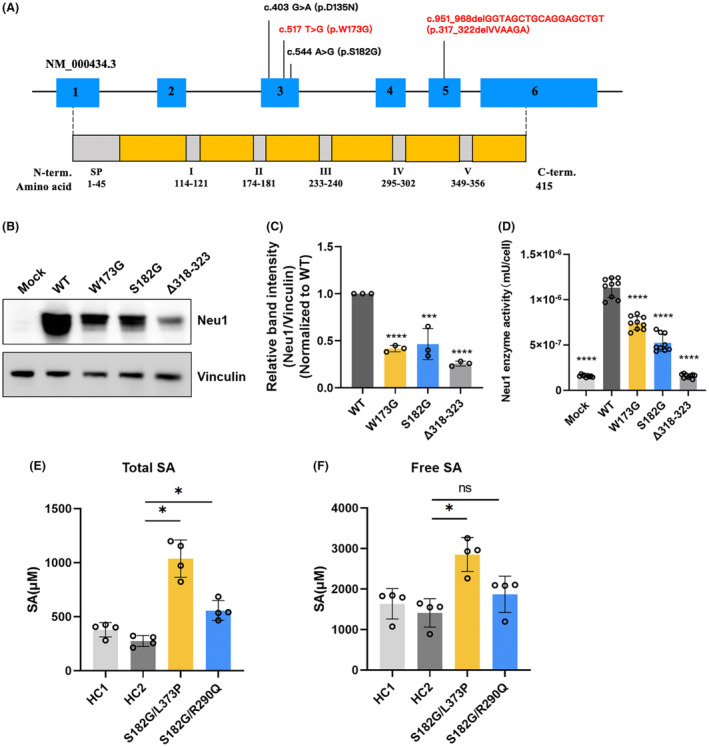
Results: A total of 31 patients from 23 unrelated families were genetically diagnosed with ST-1. A significant 80.6% of these patients were homozygous for the c.544A>G mutation. We discovered six different NEU1 variants, including two novel mutations: c.951_968del and c.517T>G. The mean age of onset was 18.0 ± 7.1 years. The clinical spectrum of ST-1 featured ataxia and myoclonus as the most common initial symptoms. Over 40% suffered from controlled generalized tonic-clonic seizures. Mobility and independence varied greatly across the cohort. Cherry-red spots were rare, occurring in just 9.5% (2/21) of patients. Brain MRIs were typically unremarkable, except for two patients with unusual findings. EEGs showed diffuse paroxysmal activity in 17 patients. The c.544A>G mutation in NEU1 is a founder variant in Fujian, with a unique haplotype prevalent in East Asians.
Interpretation: ST-1 should be suspected in patients with PMA or ataxia in Southeast China, even without macular cherry-red spots and seizures, and the premier test could be a variant screening of the founder variant NEU1 c.544A>G.
© 2024 The Author(s). Annals of Clinical and Translational Neurology published by Wiley Periodicals LLC on behalf of American Neurological Association.
Conflict of interest statement
On behalf of all authors, the corresponding author confirms no conflict of interest.
Figures
References
-
- Bonten EJ, Arts WF, Beck M, et al. Novel mutations in lysosomal neuraminidase identify functional domains and determine clinical severity in sialidosis. Hum Mol Genet. 2000;9(18):2715‐2725. - PubMed
-
- Rapin I, Goldfischer S, Katzman R, Engel J Jr, O'Brien JS. The cherry‐red spot–myoclonus syndrome. Ann Neurol. 1978;3(3):234‐242. - PubMed
-
- Lai SC, Chen RS, Wu Chou YH, et al. A longitudinal study of Taiwanese sialidosis type 1: an insight into the concept of cherry‐red spot myoclonus syndrome. Eur J Neurol. 2009;16(8):912‐919. - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
- 2020L3010/Local Science and Technology Development Project
- 82025012/National Natural Science Foundation of China
- U1905210/National Natural Science Foundation of China
- 2022YFC2703900/National Key Research and Development Program of China
- 2022YFC2703904/National Key Research and Development Program of China
- YJRC4185/Fujian Medical University Affiliated First Hospital Talent Recruitment Research Initiation
- 2021J01231/Natural Science Foundation of Fujian Province
- 2021Y9011/Joint Funds for the Innovation of Science and Technology of Fujian Province
- 2022ZD01002/Fujian Provincial Health Technology projects
- 2021ZQNZD003/Major Scientific Research Program for Middle-aged and Young Scientists of Fujian Province
LinkOut - more resources
Full Text Sources