

Inhibition of Glutamate-to-Glutathione Flux Promotes Tumor Antigen Presentation in Colorectal Cancer Cells
- PMID: 39482885
- PMCID: PMC11714253
- DOI: 10.1002/advs.202310308
Inhibition of Glutamate-to-Glutathione Flux Promotes Tumor Antigen Presentation in Colorectal Cancer Cells
Abstract
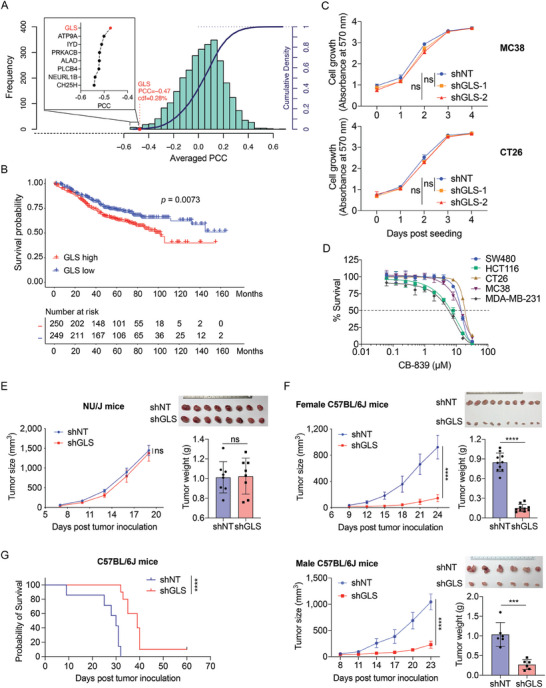
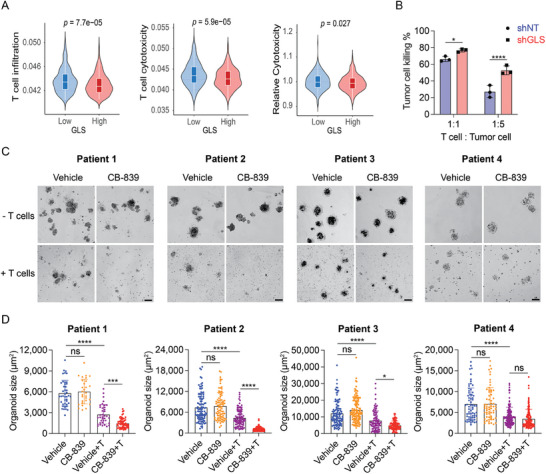
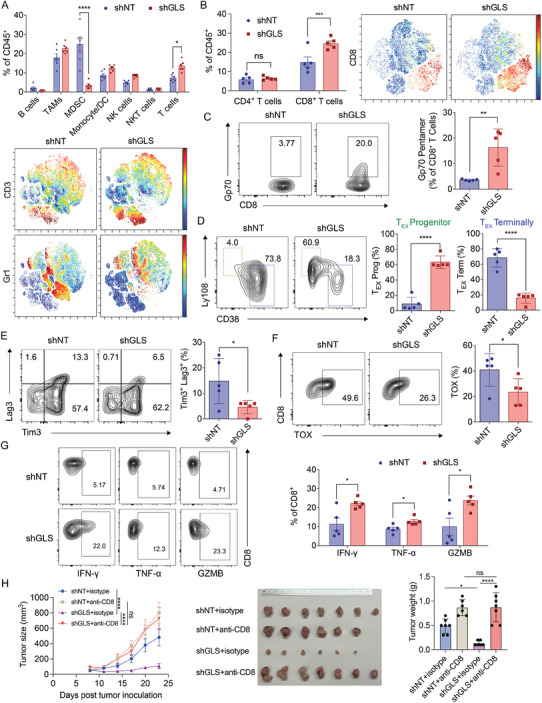
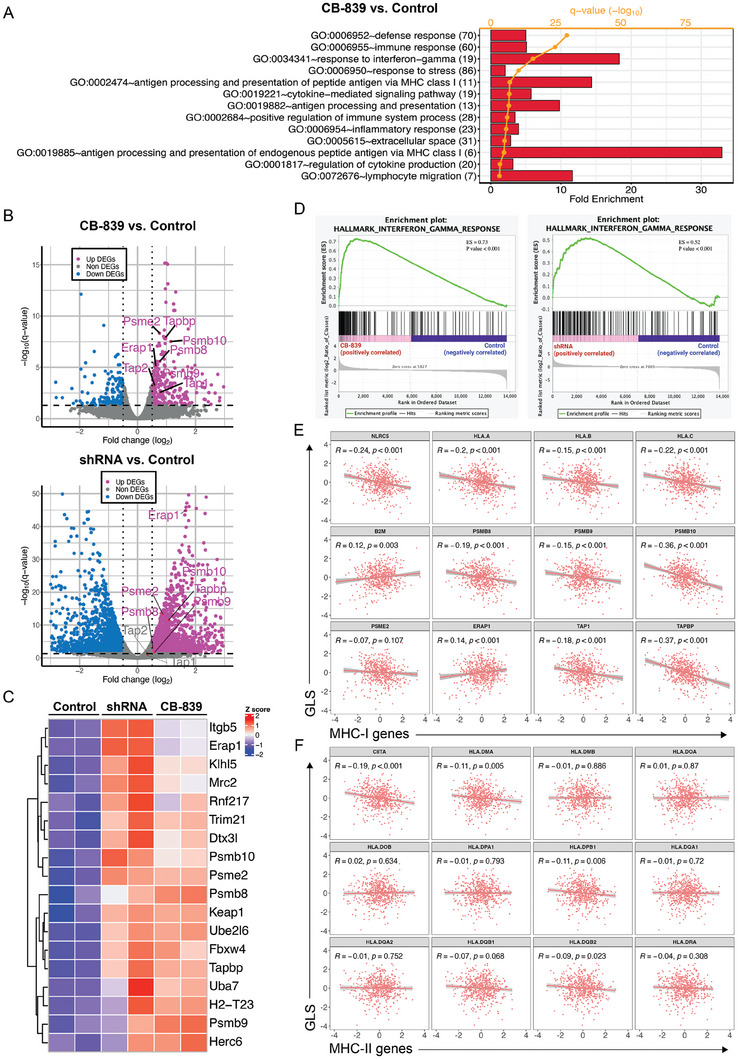
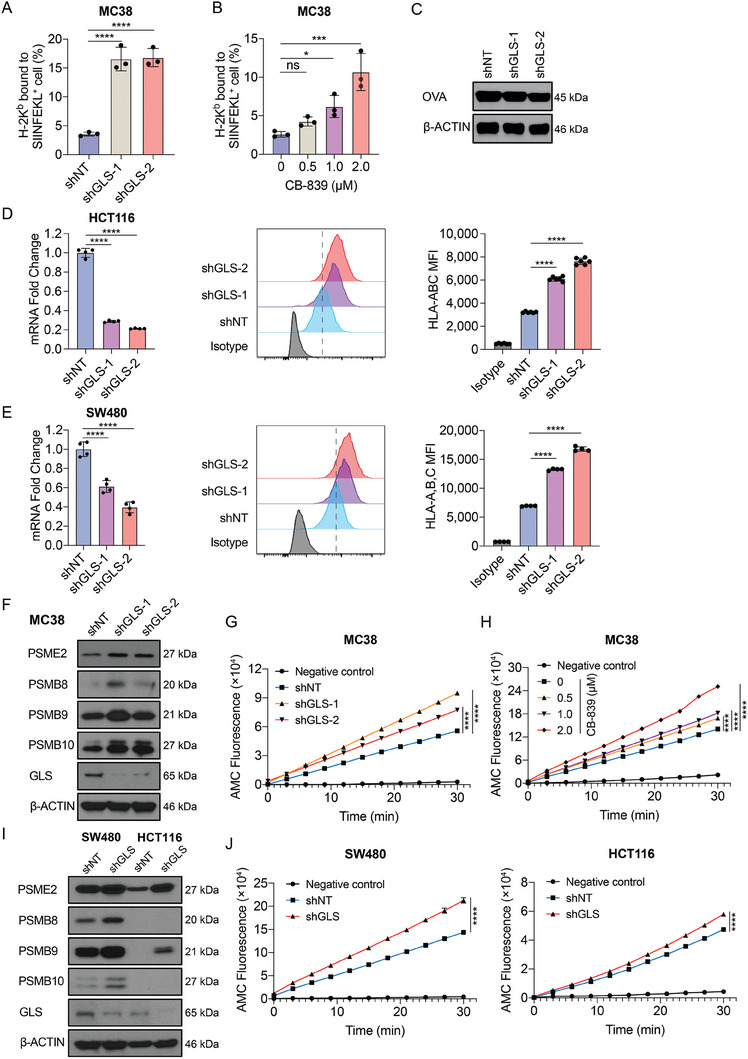
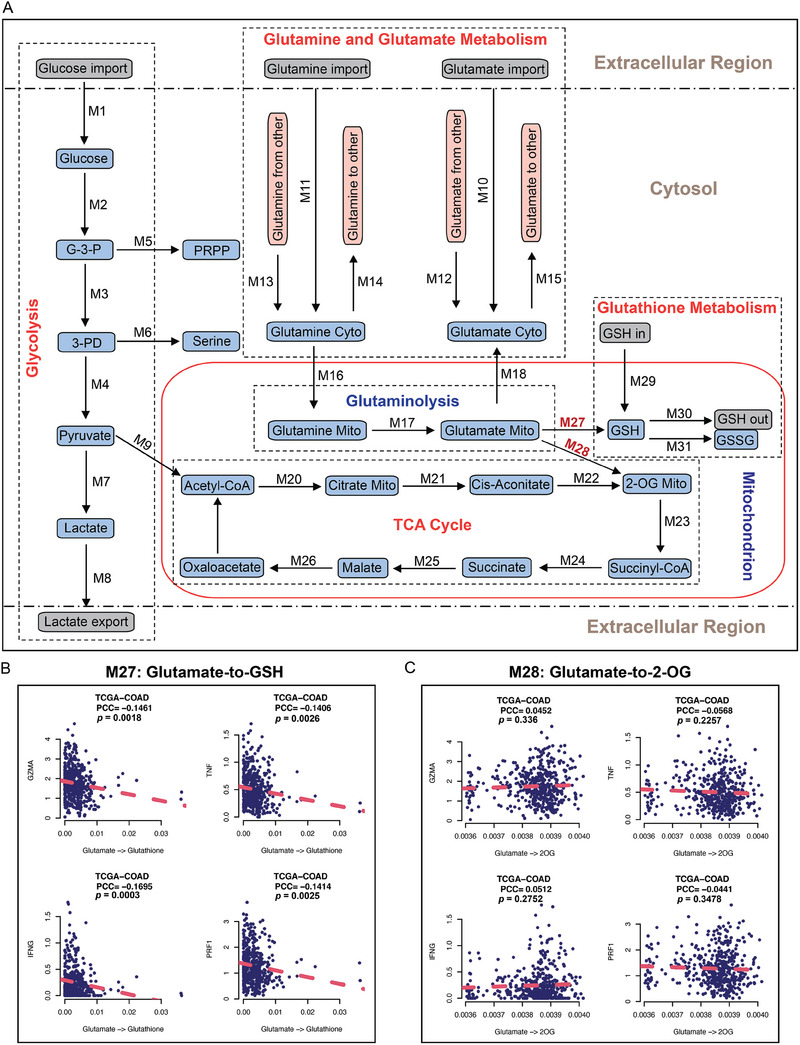
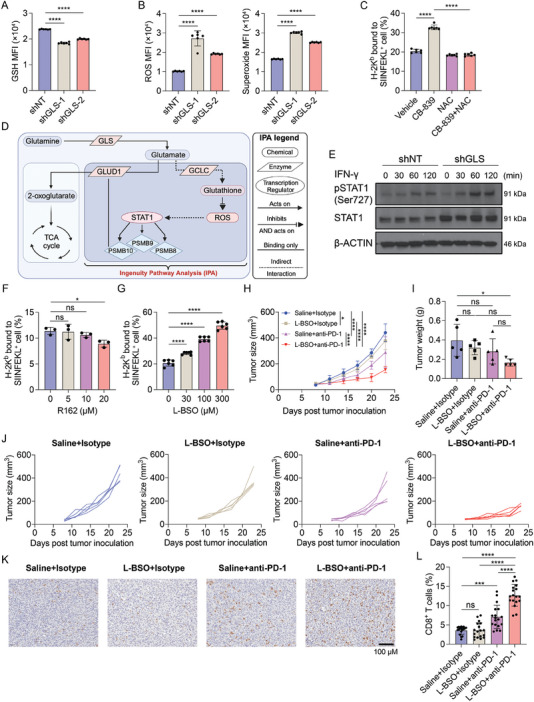
Colorectal cancer (CRC) cells display remarkable adaptability, orchestrating metabolic changes that confer growth advantages, pro-tumor microenvironment, and therapeutic resistance. One such metabolic change occurs in glutamine metabolism. Colorectal tumors with high glutaminase (GLS) expression exhibited reduced T cell infiltration and cytotoxicity, leading to poor clinical outcomes. However, depletion of GLS in CRC cells has minimal effect on tumor growth in immunocompromised mice. By contrast, remarkable inhibition of tumor growth is observed in immunocompetent mice when GLS is knocked down. It is found that GLS knockdown in CRC cells enhanced the cytotoxicity of tumor-specific T cells. Furthermore, the single-cell flux estimation analysis (scFEA) of glutamine metabolism revealed that glutamate-to-glutathione (Glu-GSH) flux, downstream of GLS, rather than Glu-to-2-oxoglutarate flux plays a key role in regulating the immune response of CRC cells in the tumor. Mechanistically, inhibition of the Glu-GSH flux activated reactive oxygen species (ROS)-related signaling pathways in tumor cells, thereby increasing the tumor immunogenicity by promoting the activity of the immunoproteasome. The combinatorial therapy of Glu-GSH flux inhibitor and anti-PD-1 antibody exhibited a superior tumor growth inhibitory effect compared to either monotherapy. Taken together, the study provides the first evidence pointing to Glu-GSH flux as a potential therapeutic target for CRC immunotherapy.
Keywords: MHC‐I antigen presentation; colorectal cancer; glutamine metabolism; immune checkpoint blockade; immunoproteasome; single‐cell flux estimation analysis.
© 2024 The Author(s). Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Siegel R. L., Wagle N. S., Cercek A., Smith R. A., Jemal A., CA Cancer J Clin. 2023, 73, 233. - PubMed
-
- a) Aparicio J., Esposito F., Serrano S., Falco E., Escudero P., Ruiz‐Casado A., Manzano H., Fernandez‐Montes A., J Clin Med. 2020, 9, 3889; - PMC - PubMed
- b) Kuipers E. J., Grady W. M., Lieberman D., Seufferlein T., Sung J. J., Boelens P. G., van de Velde C. J., Watanabe T., Nat. Rev. Dis. Primers 2015, 1, 15065. - PMC - PubMed
-
- Marcus L., Lemery S. J., Keegan P., Pazdur R., Clin. Cancer Res. 2019, 25, 3753. - PubMed
-
- Jhunjhunwala S., Hammer C., Delamarre L., Nat. Rev. Cancer 2021, 21, 298. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous