

This is a preprint.
A genome-to-proteome atlas charts natural variants controlling proteome diversity and forecasts their fitness effects
- PMID: 39484408
- PMCID: PMC11526991
- DOI: 10.1101/2024.10.18.619054
A genome-to-proteome atlas charts natural variants controlling proteome diversity and forecasts their fitness effects
Abstract
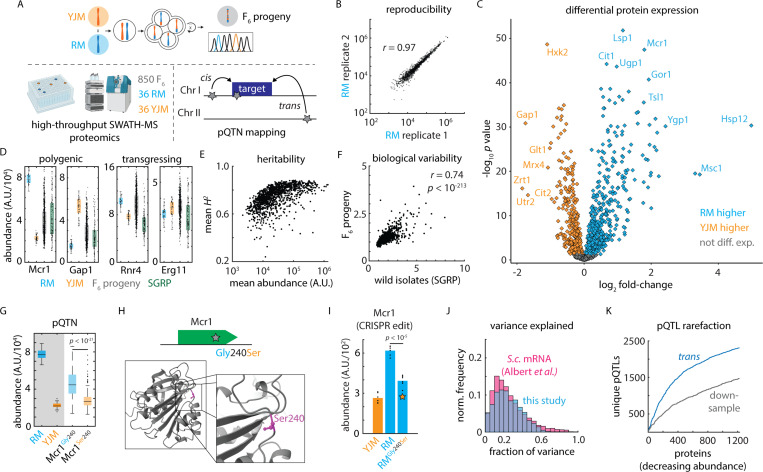
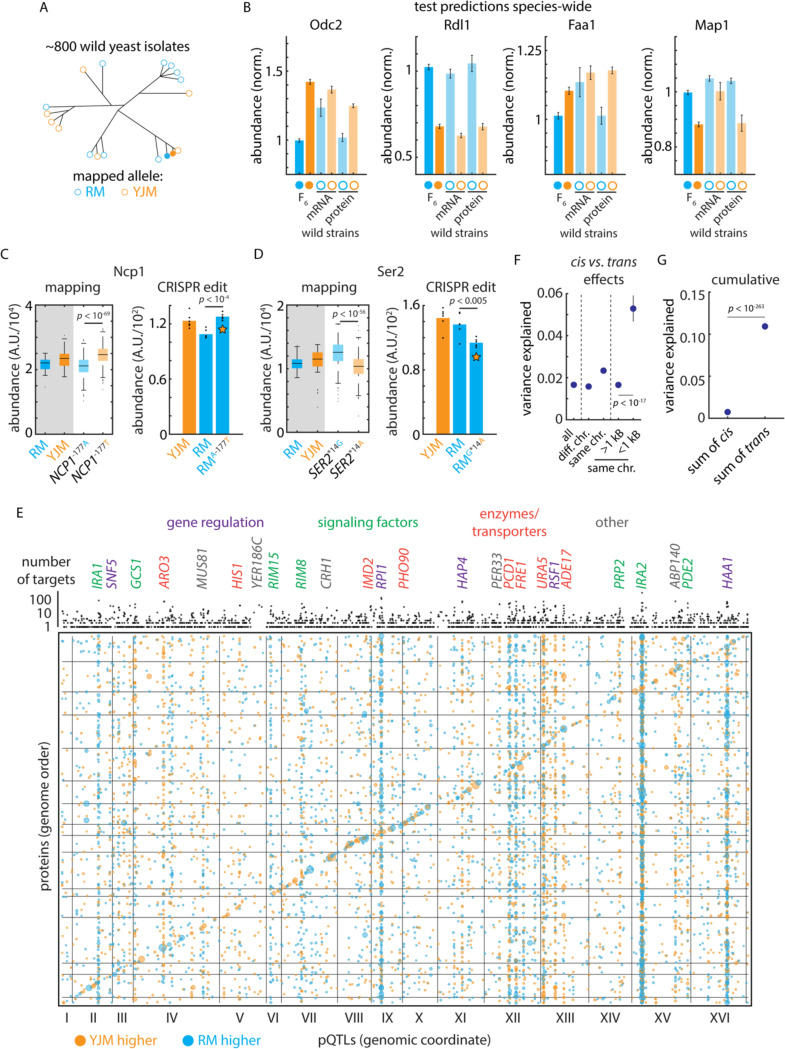
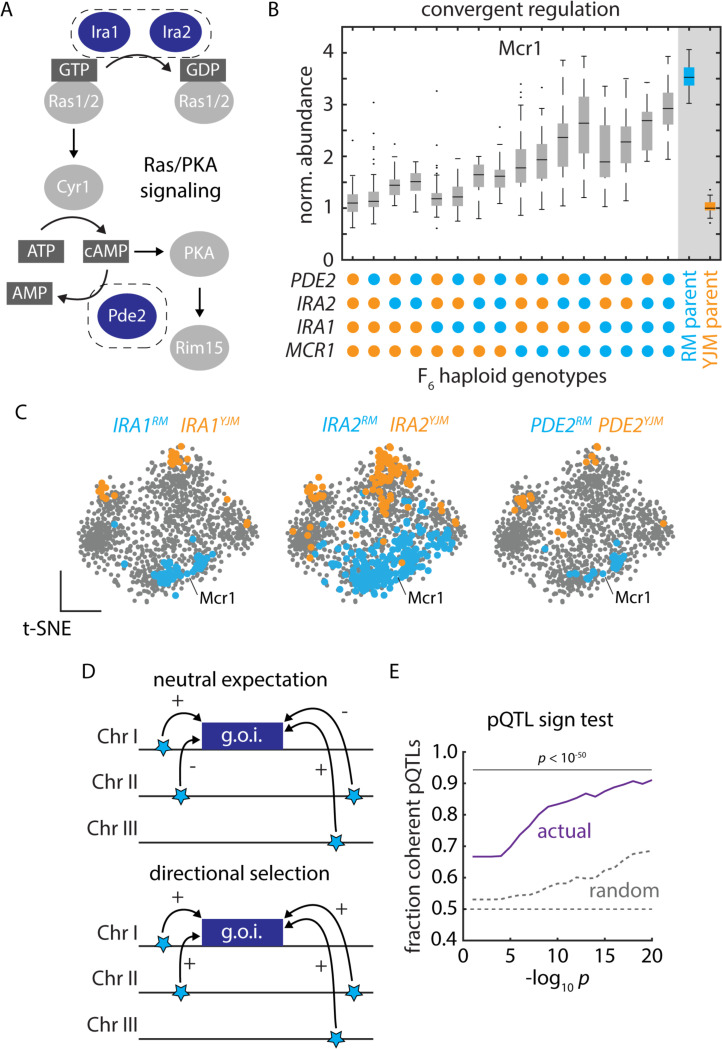
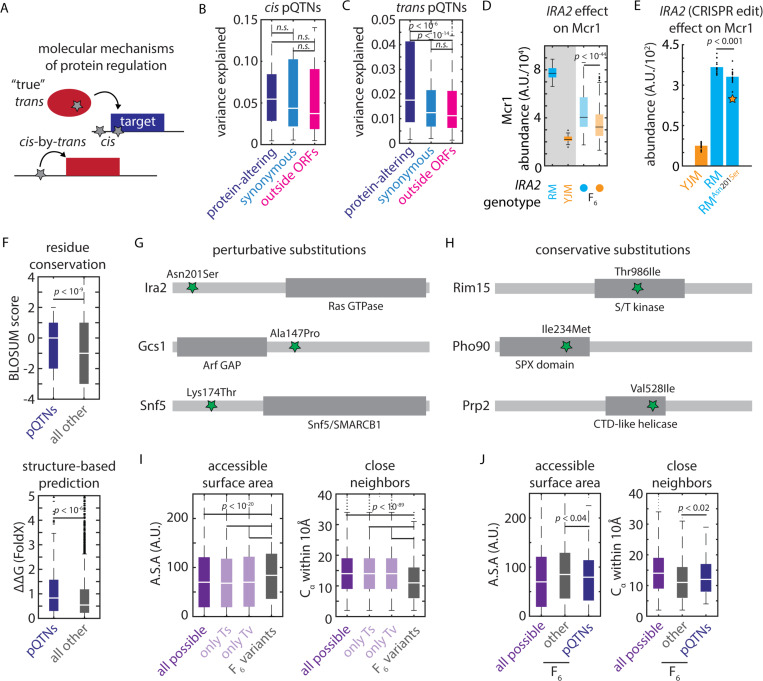
Despite abundant genomic and phenotypic data across individuals and environments, the functional impact of most mutations on phenotype remains unclear. Here, we bridge this gap by linking genome to proteome in 800 meiotic progeny from an intercross between two closely related Saccharomyces cerevisiae isolates adapted to distinct niches. Modest genetic distance between the parents generated remarkable proteomic diversity that was amplified in the progeny and captured by 6,476 genotype-protein associations, over 1,600 of which we resolved to single variants. Proteomic adaptation emerged through the combined action of numerous cis- and trans-regulatory mutations, a regulatory architecture that was conserved across the species. Notably, trans-regulatory variants often arose in proteins not traditionally associated with gene regulation, such as enzymes. Moreover, the proteomic consequences of mutations predicted fitness under various stresses. Our study demonstrates that the collective action of natural genetic variants drives dramatic proteome diversification, with molecular consequences that forecast phenotypic outcomes.
Keywords: adaptation; gene expression evolution; genotype-phenotype mapping; omnigenic model; proteomics; systems genetics; transgression; variant interpretation.
Conflict of interest statement
Declaration of Interests M. Ralser is founder and shareholder of Eliptica Ltd. The other authors declare no competing interests.
Figures
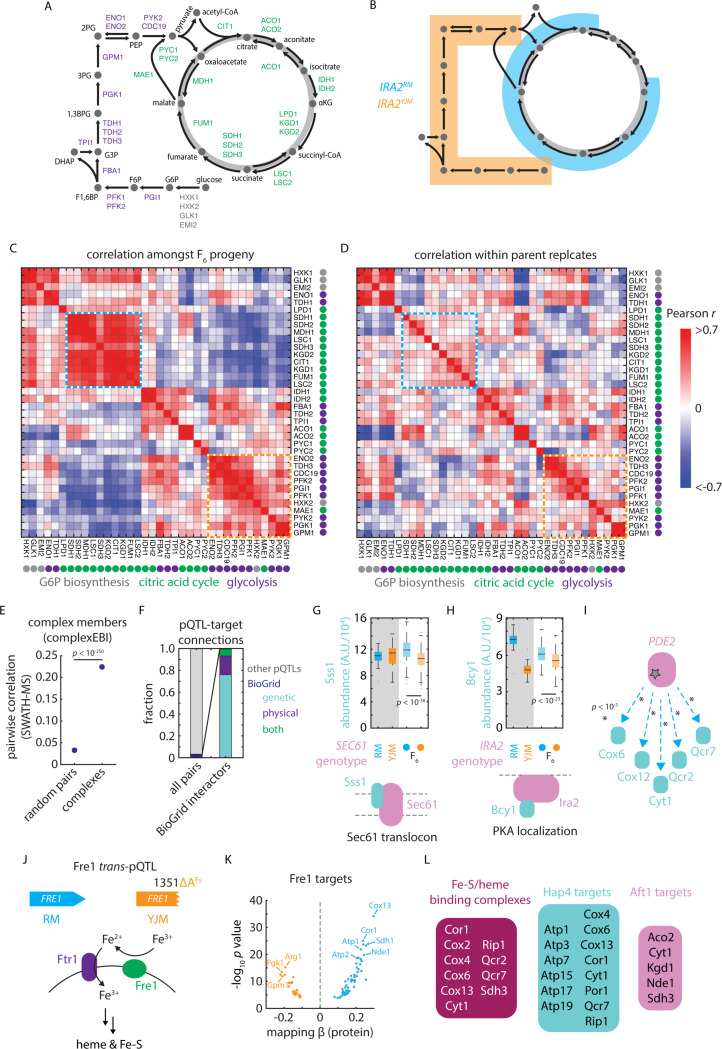
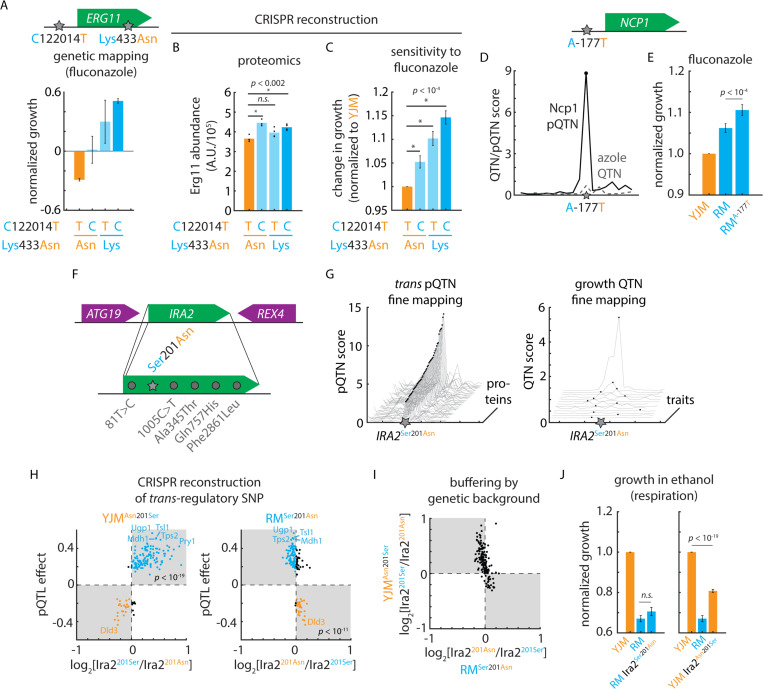
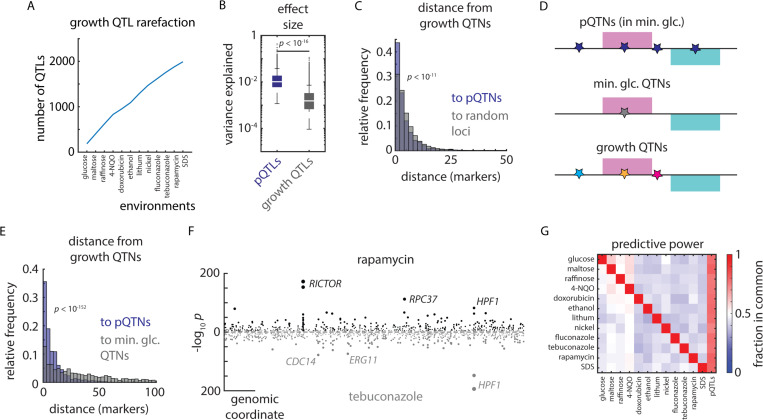
References
-
- Leiding J.W., Vogel T.P., Santarlas V.G.J., Mhaskar R., Smith M.R., Carisey A., Vargas-Hernández A., Silva-Carmona M., Heeg M., Rensing-Ehl A., et al. (2023). Monogenic early-onset lymphoproliferation and autoimmunity: Natural history of STAT3 gain-of-function syndrome. J. Allergy Clin. Immunol. 151, 1081–1095. - PMC - PubMed
-
- Wagner N., Çelik M.H., Hölzlwimmer F.R., Mertes C., Prokisch H., Yépez V.A., and Gagneur J. (2023). Aberrant splicing prediction across human tissues. Nat. Genet. 55, 861–870. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous