

This is a preprint.
An integrated view of the structure and function of the human 4D nucleome
- PMID: 39484446
- PMCID: PMC11526861
- DOI: 10.1101/2024.09.17.613111
An integrated view of the structure and function of the human 4D nucleome
Update in
-
An integrated view of the structure and function of the human 4D nucleome.Nature. 2026 Jan;649(8097):759-776. doi: 10.1038/s41586-025-09890-3. Epub 2025 Dec 17. Nature. 2026. PMID: 41407856 Free PMC article.
Abstract
The dynamic three-dimensional (3D) organization of the human genome (the "4D Nucleome") is closely linked to genome function. Here, we integrate a wide variety of genomic data generated by the 4D Nucleome Project to provide a detailed view of human 3D genome organization in widely used embryonic stem cells (H1-hESCs) and immortalized fibroblasts (HFFc6). We provide extensive benchmarking of 3D genome mapping assays and integrate these diverse datasets to annotate spatial genomic features across scales. The data reveal a rich complexity of chromatin domains and their sub-nuclear positions, and over one hundred thousand structural loops and promoter-enhancer interactions. We developed 3D models of population-based and individual cell-to-cell variation in genome structure, establishing connections between chromosome folding, nuclear organization, chromatin looping, gene transcription, and DNA replication. We demonstrate the use of computational methods to predict genome folding from DNA sequence, uncovering potential effects of genetic variants on genome structure and function. Together, this comprehensive analysis contributes insights into human genome organization and enhances our understanding of connections between the regulation of genome function and 3D genome organization in general.
Conflict of interest statement
Conflicts of interest Job Dekker is a member of the scientific advisory board of Arima Genomics, San Diego, CA, USA and Omega Therapeutic, Cambridge, MA, USA. Sheng Zhong. is a founder and shareholder of Genemo, Inc., San Diego, CA, USA Bing Ren has equity in Arima Genomics Inc., San Diego, CA, USA and Epigenome Technologies, San Diego, CA, USA Clair Marchal is the director and founder of In silichrom ltd.
Figures








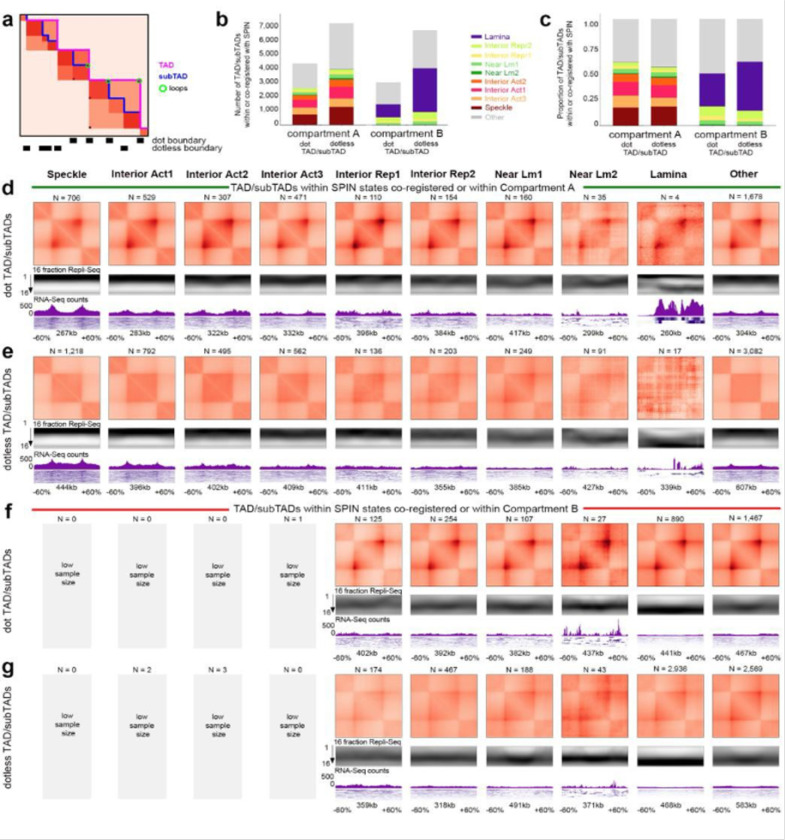

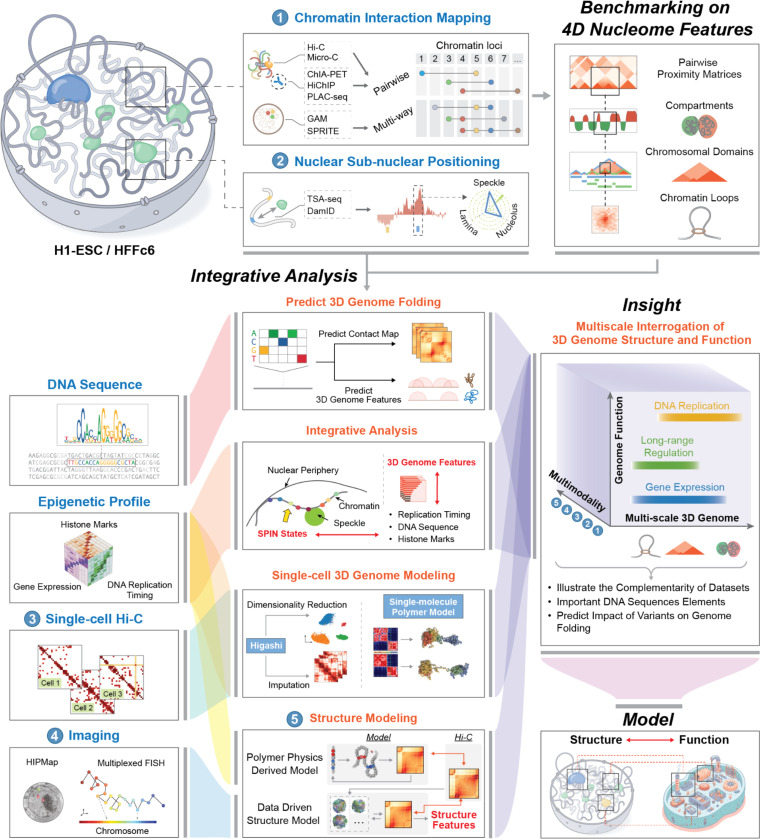








References
Publication types
Grants and funding
- U54 DK107981/DK/NIDDK NIH HHS/United States
- T32 GM008042/GM/NIGMS NIH HHS/United States
- U54 DK107977/DK/NIDDK NIH HHS/United States
- U54 DK107967/DK/NIDDK NIH HHS/United States
- U01 DA052715/DA/NIDA NIH HHS/United States
- U01 CA200060/CA/NCI NIH HHS/United States
- U54 DK107979/DK/NIDDK NIH HHS/United States
- U01 DK127405/DK/NIDDK NIH HHS/United States
- U01 HL129998/HL/NHLBI NIH HHS/United States
- U01 CA200147/CA/NCI NIH HHS/United States
- UM1 HG011593/HG/NHGRI NIH HHS/United States
- U01 HL130007/HL/NHLBI NIH HHS/United States
- UM1 HG011536/HG/NHGRI NIH HHS/United States
- U54 DK107980/DK/NIDDK NIH HHS/United States
- U01 DA052769/DA/NIDA NIH HHS/United States
- U01 DA040612/DA/NIDA NIH HHS/United States
- U01 DK127420/DK/NIDDK NIH HHS/United States
- U54 DK107965/DK/NIDDK NIH HHS/United States
- UM1 HG011585/HG/NHGRI NIH HHS/United States
- U01 CA200059/CA/NCI NIH HHS/United States
- U01 HL157989/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources