

This is a preprint.
Comprehensive structure-function analysis reveals gain- and loss-of-function mechanisms impacting oncogenic KRAS activity
- PMID: 39484452
- PMCID: PMC11526993
- DOI: 10.1101/2024.10.22.618529
Comprehensive structure-function analysis reveals gain- and loss-of-function mechanisms impacting oncogenic KRAS activity
Abstract
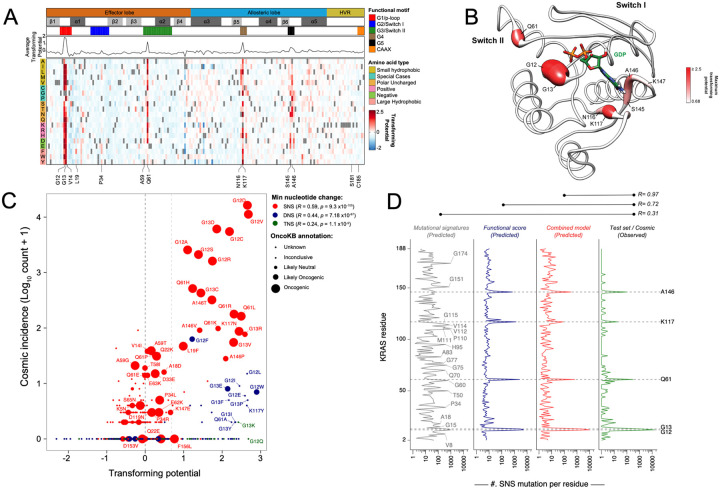
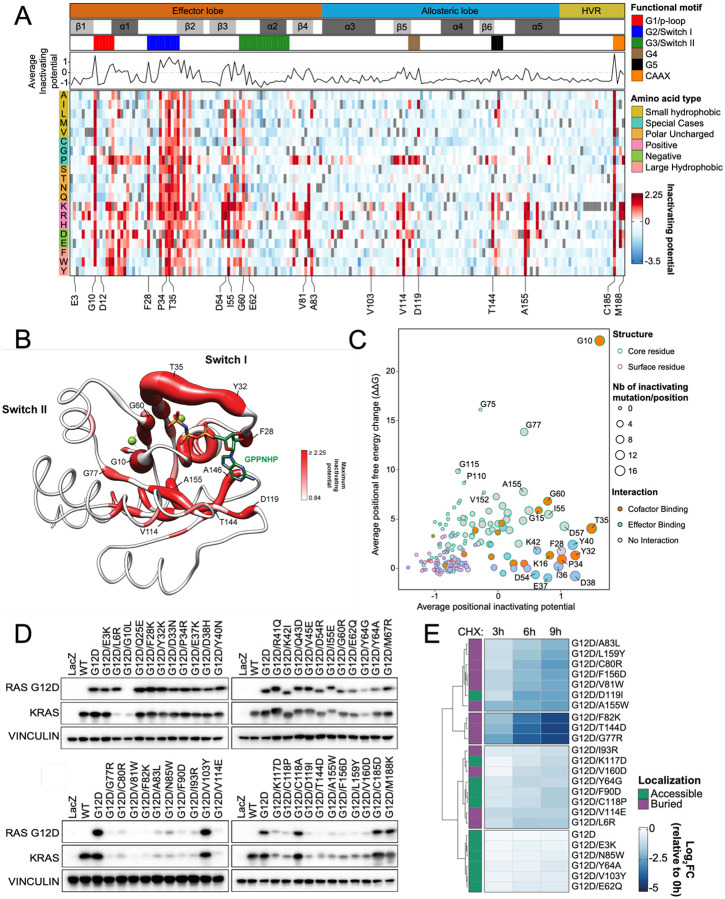
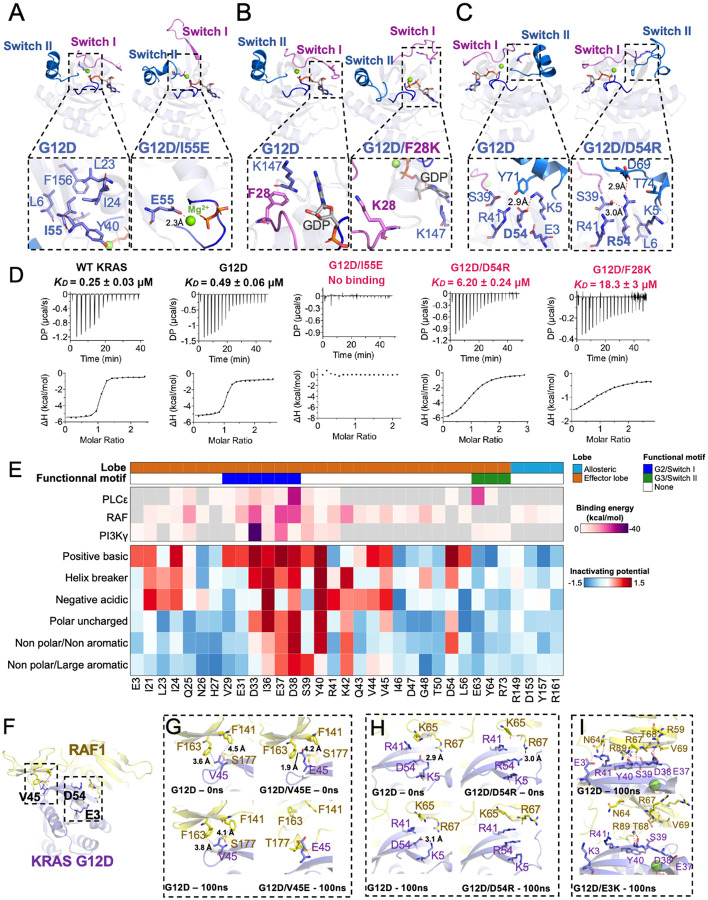
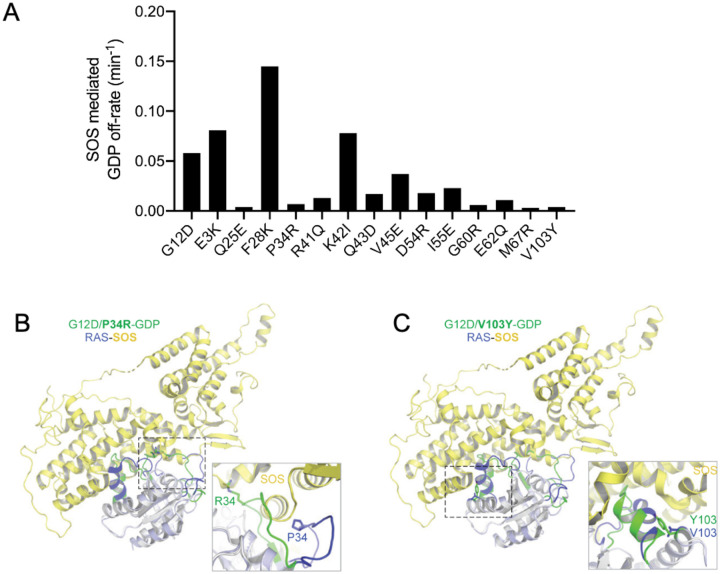
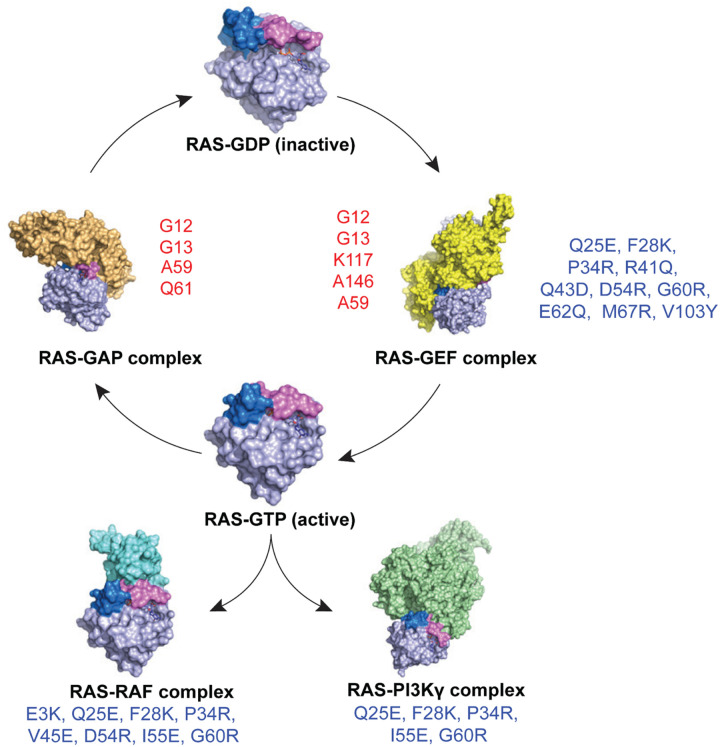
To dissect variant-function relationships in the KRAS oncoprotein, we performed deep mutational scanning (DMS) screens for both wild-type and KRASG12D mutant alleles. We defined the spectrum of oncogenic potential for nearly all possible KRAS variants, identifying several novel transforming alleles and elucidating a model to describe the frequency of KRAS mutations in human cancer as a function of transforming potential, mutational probability, and tissue-specific mutational signatures. Biochemical and structural analyses of variants identified in a KRASG12D second-site suppressor DMS screen revealed that attenuation of oncogenic KRAS can be mediated by protein instability and conformational rigidity, resulting in reduced binding affinity to effector proteins, such as RAF and PI3-kinases, or reduced SOS-mediated nucleotide exchange activity. These studies define the landscape of single amino acid alterations that modulate the function of KRAS, providing a resource for the clinical interpretation of KRAS variants and elucidating mechanisms of oncogenic KRAS inactivation for therapeutic exploitation.
Keywords: KRAS; cancers; cell transformation; deep mutational scanning; structure-function.
Figures
References
-
- Hingorani S. R. et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483 (2005). - PubMed
-
- Ji H. et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 448, 807–810 (2007). - PubMed
-
- Brummelkamp T. R., Bernards R. & Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science 296, 550–553 (2002). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous