

Leveraging the T2T assembly to resolve rare and pathogenic inversions in reference genome gaps
- PMID: 39486878
- PMCID: PMC11610578
- DOI: 10.1101/gr.279346.124
Leveraging the T2T assembly to resolve rare and pathogenic inversions in reference genome gaps
Abstract
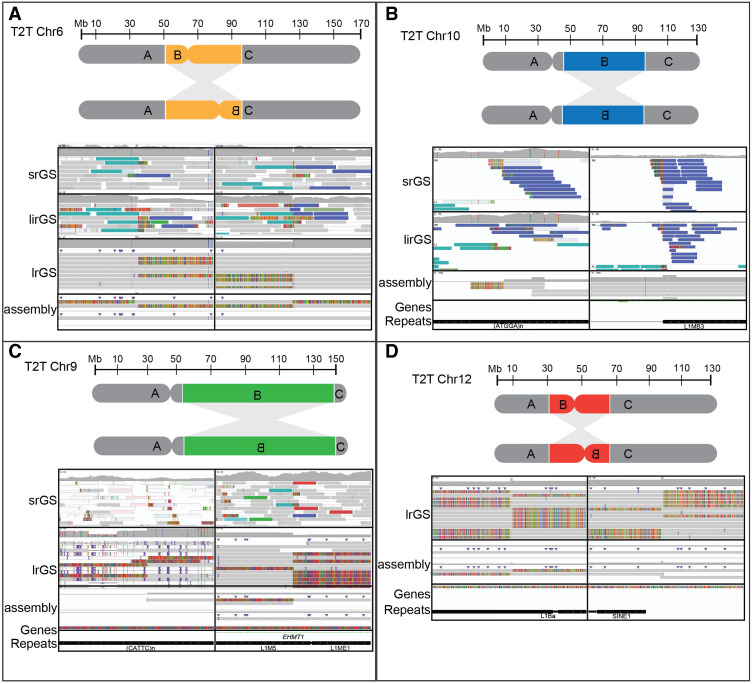
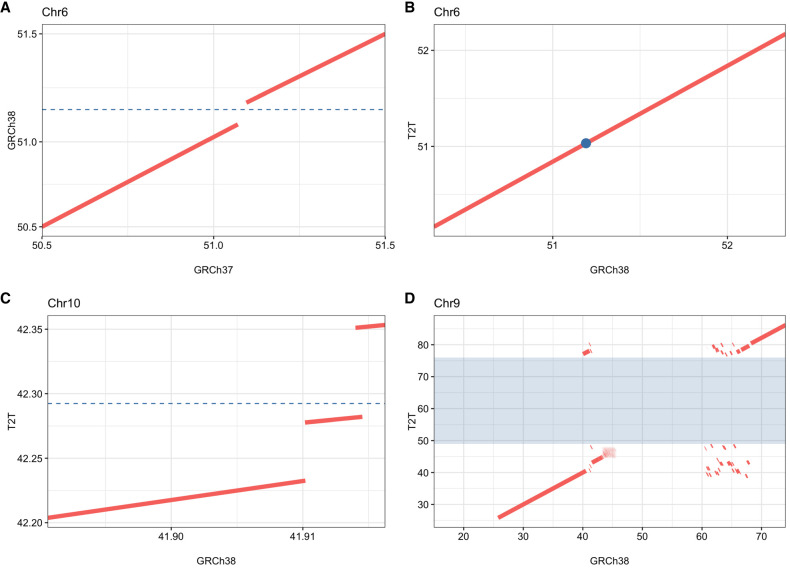
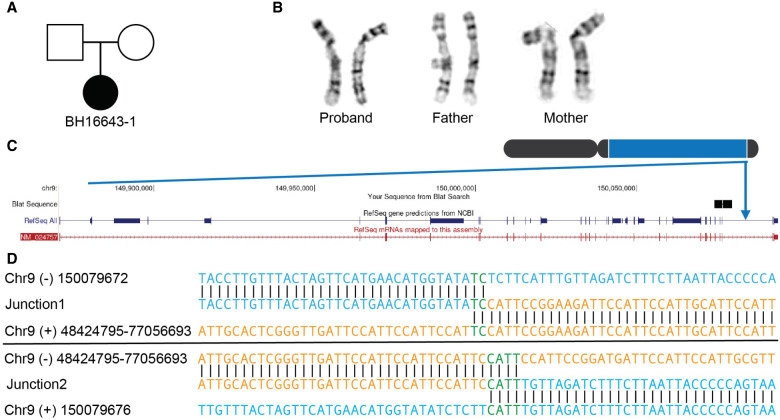
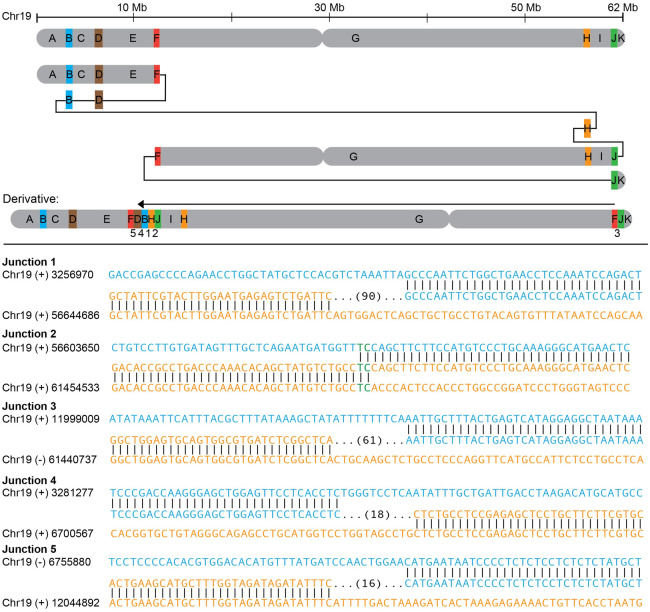
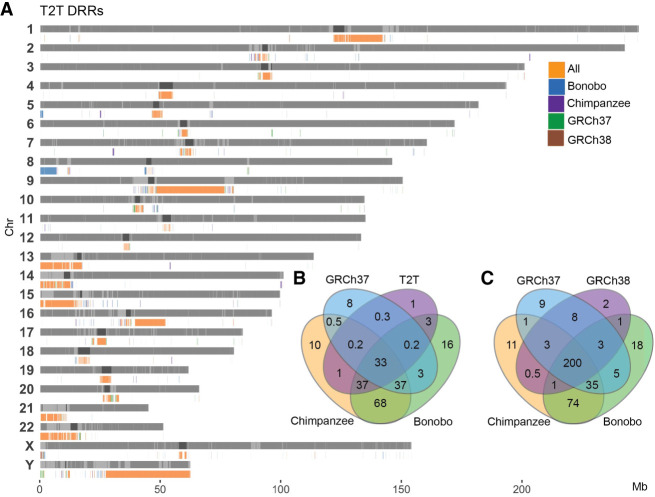
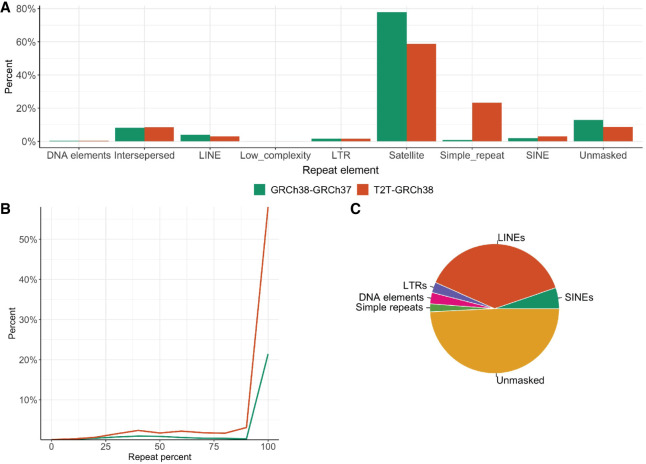
Chromosomal inversions (INVs) are particularly challenging to detect due to their copy-number neutral state and association with repetitive regions. Inversions represent about 1/20 of all balanced structural chromosome aberrations and can lead to disease by gene disruption or altering regulatory regions of dosage-sensitive genes in cis Short-read genome sequencing (srGS) can only resolve ∼70% of cytogenetically visible inversions referred to clinical diagnostic laboratories, likely due to breakpoints in repetitive regions. Here, we study 12 inversions by long-read genome sequencing (lrGS) (n = 9) or srGS (n = 3) and resolve nine of them. In four cases, the inversion breakpoint region was missing from at least one of the human reference genomes (GRCh37, GRCh38, T2T-CHM13) and a reference agnostic analysis was needed. One of these cases, an INV9 mappable only in de novo assembled lrGS data using T2T-CHM13 disrupts EHMT1 consistent with a Mendelian diagnosis (Kleefstra syndrome 1; MIM#610253). Next, by pairwise comparison between T2T-CHM13, GRCh37, and GRCh38, as well as the chimpanzee and bonobo, we show that hundreds of megabases of sequence are missing from at least one human reference, highlighting that primate genomes contribute to genomic diversity. Aligning population genomic data to these regions indicated that these regions are variable between individuals. Our analysis emphasizes that T2T-CHM13 is necessary to maximize the value of lrGS for optimal inversion detection in clinical diagnostics. These results highlight the importance of leveraging diverse and comprehensive reference genomes to resolve unsolved molecular cases in rare diseases.
© 2024 Bilgrav Saether et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Update of
-
Mind the gap: the relevance of the genome reference to resolve rare and pathogenic inversions.medRxiv [Preprint]. 2024 Apr 24:2024.04.22.24305780. doi: 10.1101/2024.04.22.24305780. medRxiv. 2024. Update in: Genome Res. 2024 Nov 20;34(11):1785-1797. doi: 10.1101/gr.279346.124. PMID: 38712270 Free PMC article. Updated. Preprint.
References
-
- Ameur A, Che H, Martin M, Bunikis I, Dahlberg J, Höijer I, Häggqvist S, Vezzi F, Nordlund J, Olason P, et al. 2018. De novo assembly of two Swedish genomes reveals missing segments from the human GRCh38 reference and improves variant calling of population-scale sequencing data. Genes (Basel) 9: 486. 10.3390/genes9100486 - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources