

HiDDEN: a machine learning method for detection of disease-relevant populations in case-control single-cell transcriptomics data
- PMID: 39487129
- PMCID: PMC11530671
- DOI: 10.1038/s41467-024-53666-8
HiDDEN: a machine learning method for detection of disease-relevant populations in case-control single-cell transcriptomics data
Abstract
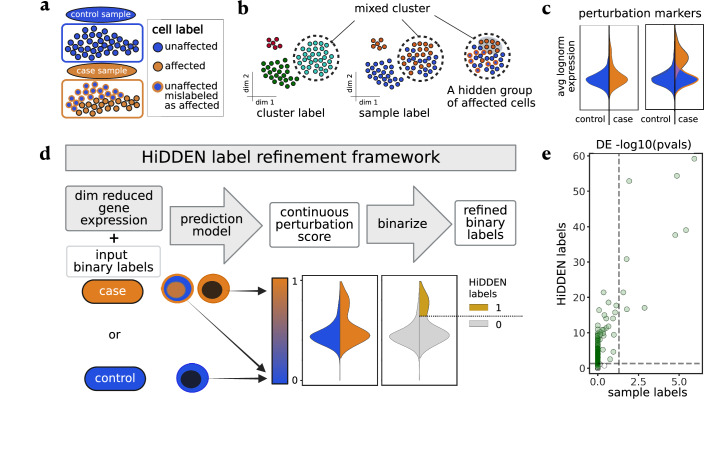
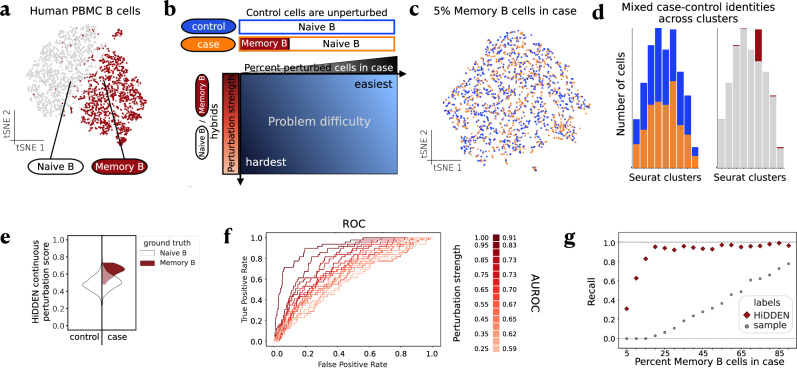
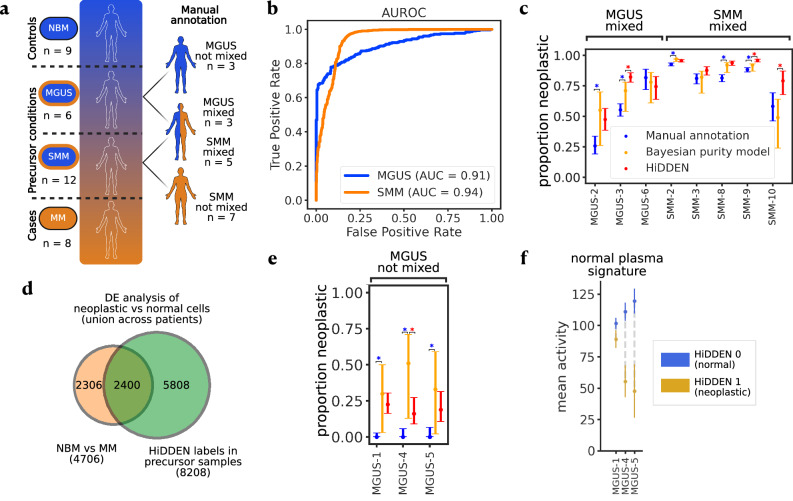
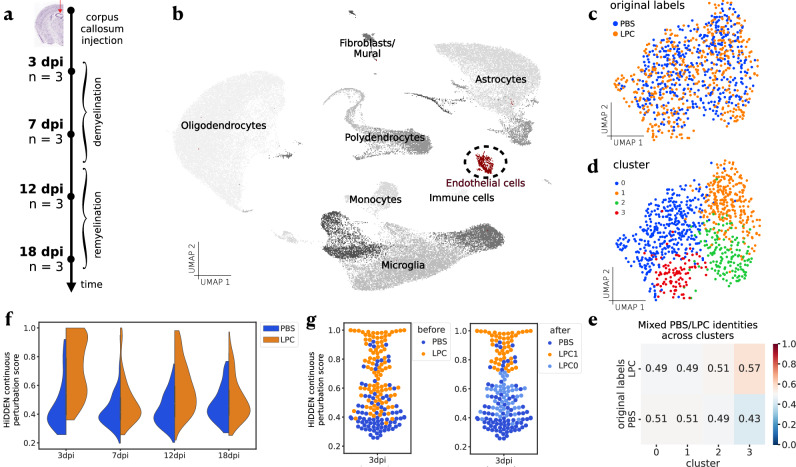
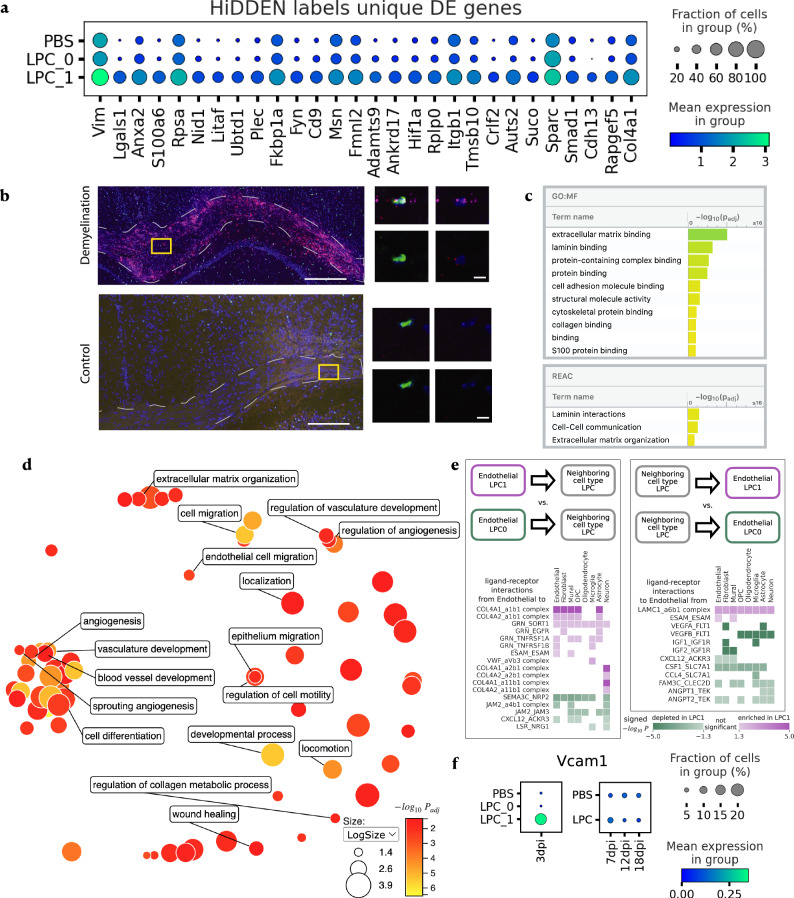
In case-control single-cell RNA-seq studies, sample-level labels are transferred onto individual cells, labeling all case cells as affected, when in reality only a small fraction of them may actually be perturbed. Here, using simulations, we demonstrate that the standard approach to single cell analysis fails to isolate the subset of affected case cells and their markers when either the affected subset is small, or when the strength of the perturbation is mild. To address this fundamental limitation, we introduce HiDDEN, a computational method that refines the case-control labels to accurately reflect the perturbation status of each cell. We show HiDDEN's superior ability to recover biological signals missed by the standard analysis workflow in simulated ground truth datasets of cell type mixtures. When applied to a dataset of human multiple myeloma precursor conditions, HiDDEN recapitulates the expert manual annotation and discovers malignancy in early stage samples missed in the original analysis. When applied to a mouse model of demyelination, HiDDEN identifies an endothelial subpopulation playing a role in early stage blood-brain barrier dysfunction. We anticipate that HiDDEN should find wide usage in contexts that require the detection of subtle transcriptional changes in cell types across conditions.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Kamath, T. et al. A molecular census of midbrain dopaminergic neurons in Parkinson’s disease. bioRxiv10.1101/2021.06.16.448661 (2021).
Publication types
MeSH terms
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
