

Oncogenic EML4-ALK assemblies suppress growth factor perception and modulate drug tolerance
- PMID: 39488530
- PMCID: PMC11531495
- DOI: 10.1038/s41467-024-53451-7
Oncogenic EML4-ALK assemblies suppress growth factor perception and modulate drug tolerance
Abstract
Drug resistance remains a challenge for targeted therapy of cancers driven by EML4-ALK and related fusion oncogenes. EML4-ALK forms cytoplasmic protein condensates, which result from networks of interactions between oncogene and adapter protein multimers. While these assemblies are associated with oncogenic signaling, their role in drug response is unclear. Here, we use optogenetics and live-cell imaging to find that EML4-ALK assemblies suppress transmembrane receptor tyrosine kinase (RTK) signaling by sequestering RTK adapter proteins including GRB2 and SOS1. Furthermore, ALK inhibition, while suppressing oncogenic signaling, simultaneously releases the sequestered adapters and thereby resensitizes RTK signaling. Resensitized RTKs promote rapid and pulsatile ERK reactivation that originates from paracrine ligands shed by dying cells. Reactivated ERK signaling promotes cell survival, which can be counteracted by combination therapies that block paracrine signaling. Our results identify a regulatory role for RTK fusion assemblies and uncover a mechanism of tolerance to targeted therapies.
© 2024. The Author(s).
Conflict of interest statement
D.G.M. and L.J.B. have filed a provisional patent based on the findings in this work. T.G.B. is an advisor to Novartis, AstraZeneca, Revolution Medicines, Array/Pfizer, Springworks, Strategia, Relay, Jazz, Rain, Engine, Scorpion and receives research funding from Strategia, Kinnate, Verastem, and Revolution Medicines. A.T. is an advisor to Faze Medicines. R.C.D. is an employee and shareholder of Rain Oncology Inc. and has received licensing fees from Takeda, ThermoFisher, Voronoi, Loxo, Histocyte, and Black Diamond. The remaining authors declare no competing interests.
Figures


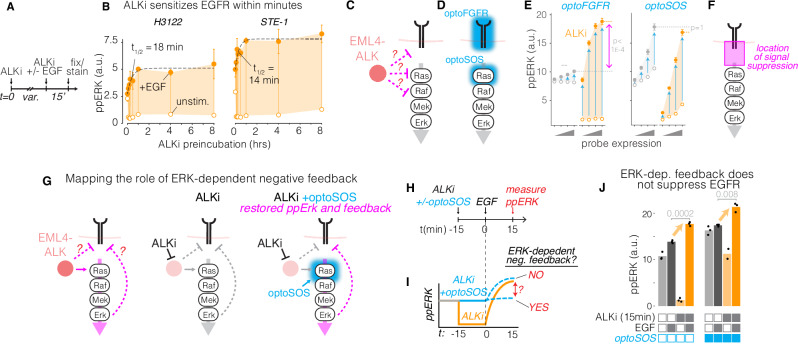
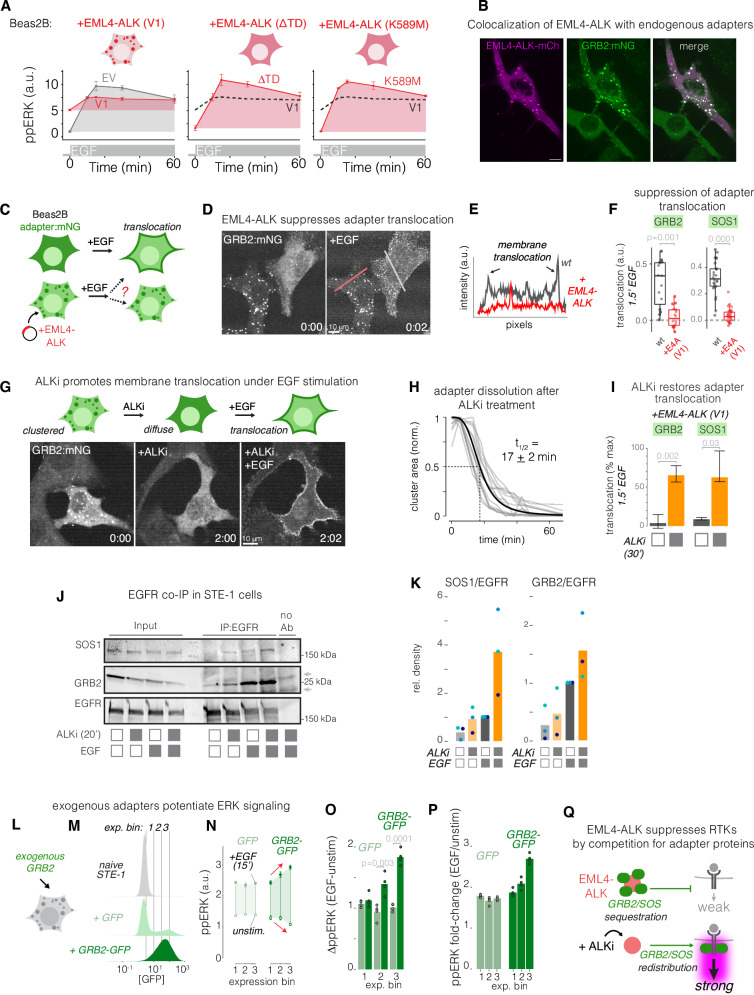
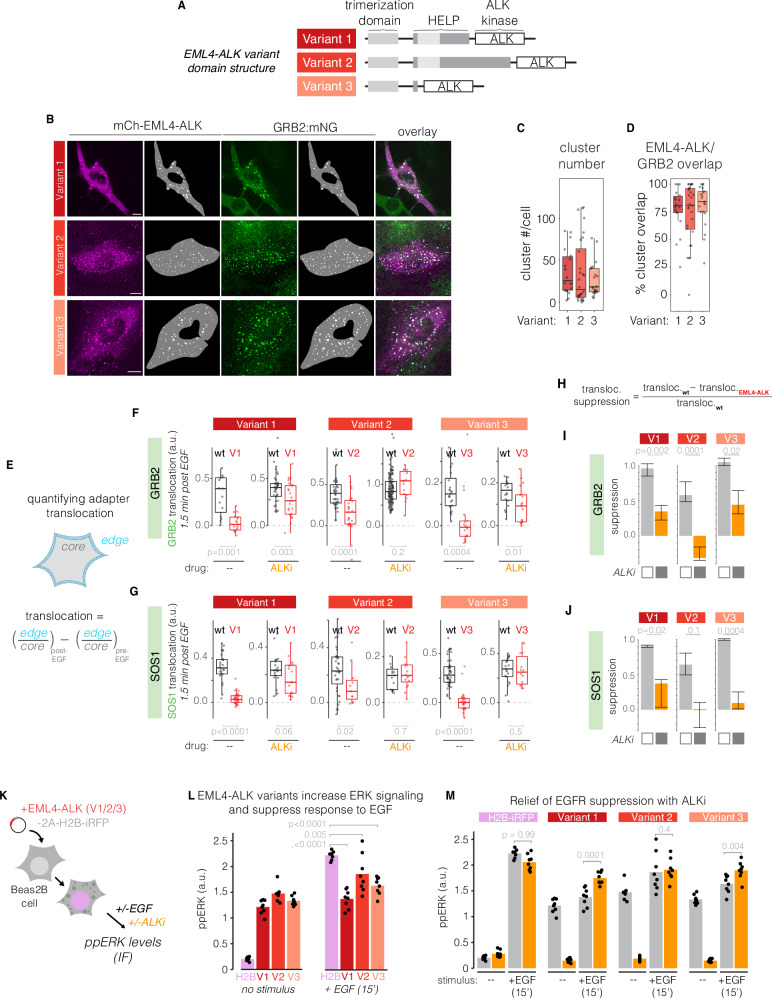
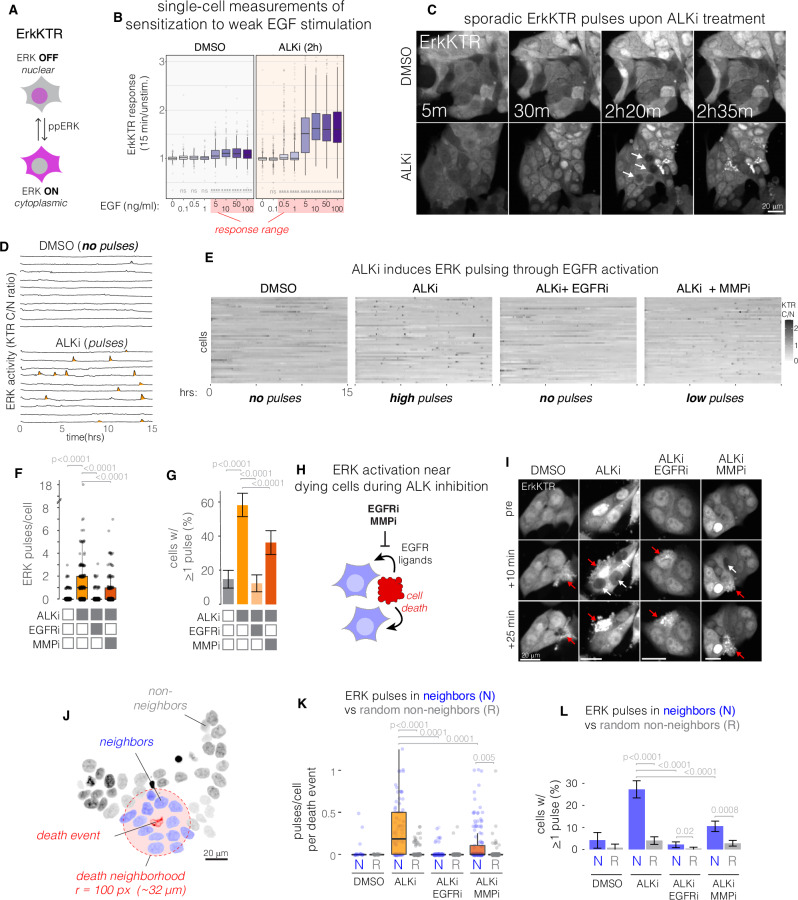

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
