

Genomes and epigenomes of matched normal and tumor breast tissue reveal diverse evolutionary trajectories and tumor-host interactions
- PMID: 39492056
- PMCID: PMC11639081
- DOI: 10.1016/j.ajhg.2024.10.005
Genomes and epigenomes of matched normal and tumor breast tissue reveal diverse evolutionary trajectories and tumor-host interactions
Abstract
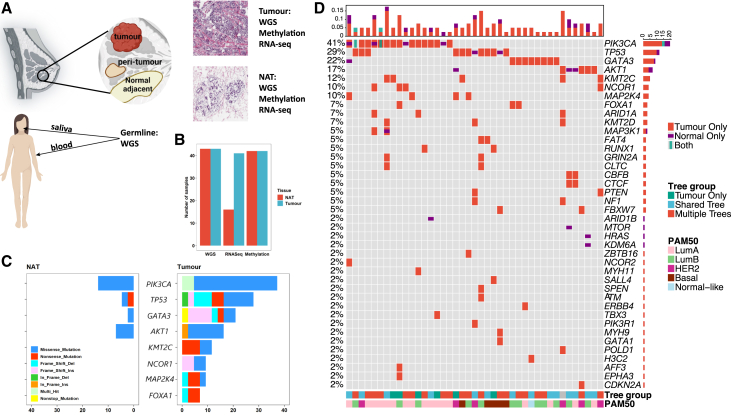
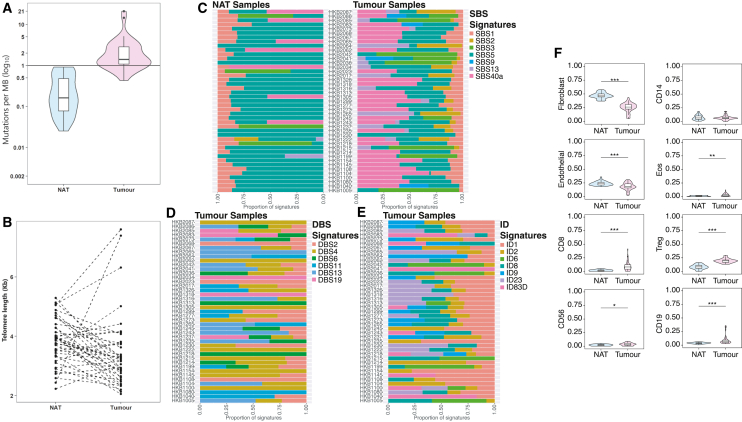
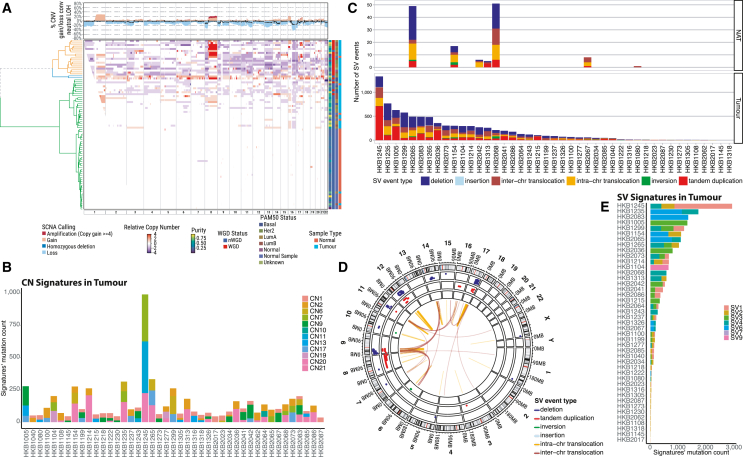
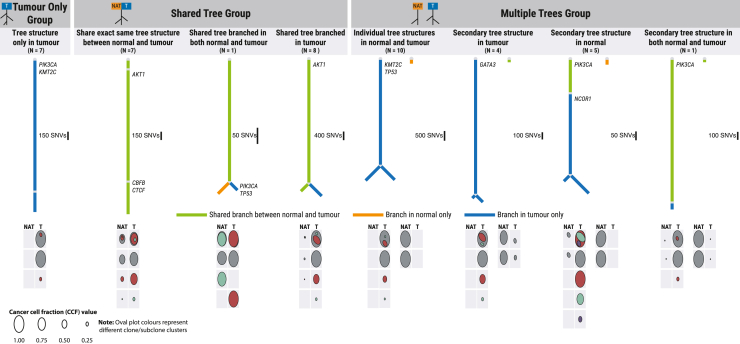
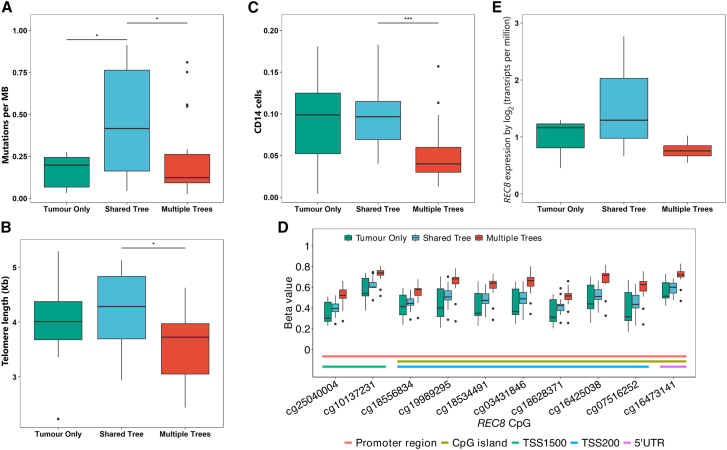
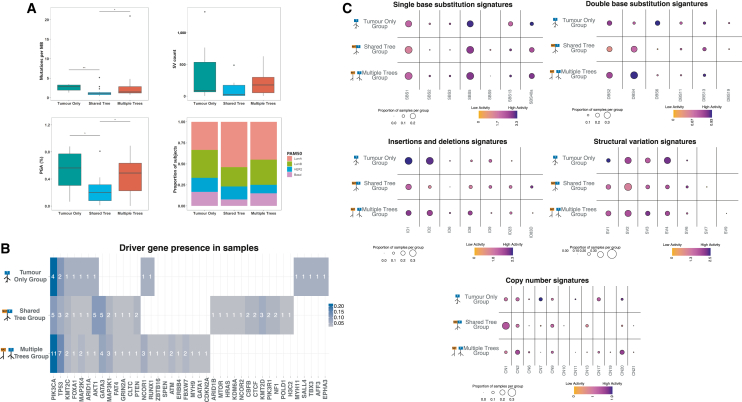
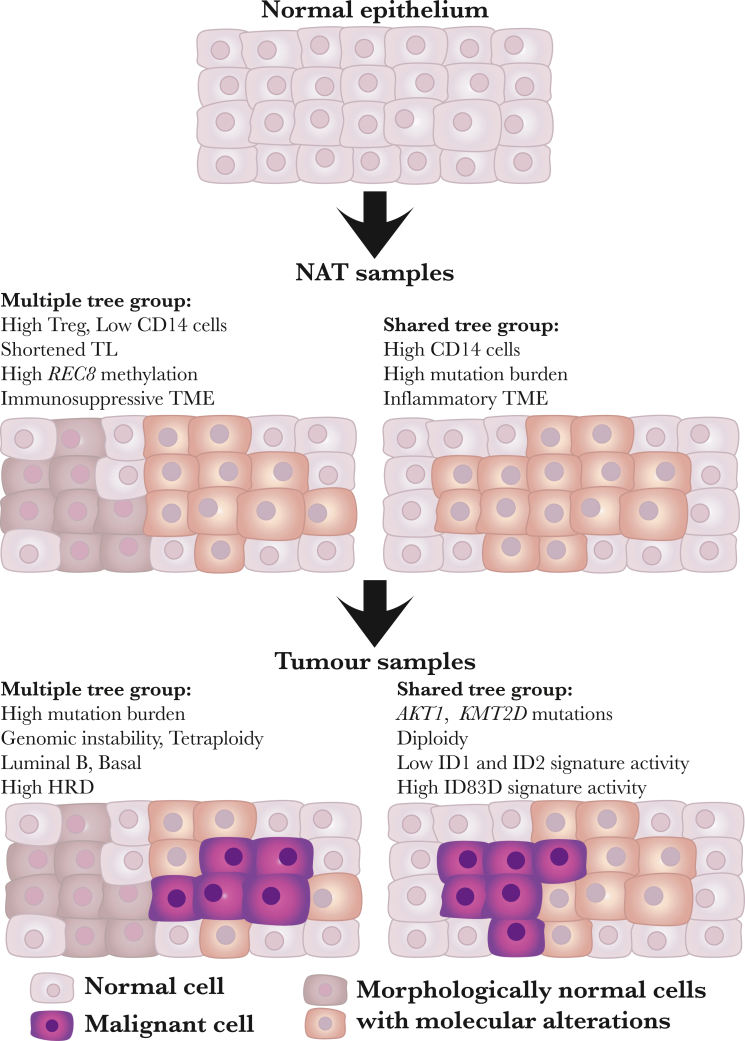
Normal tissues adjacent to the tumor (NATs) may harbor early breast carcinogenesis events driven by field cancerization. Although previous studies have characterized copy-number (CN) and transcriptomic alterations, the evolutionary history of NATs in breast cancer (BC) remains poorly characterized. Utilizing whole-genome sequencing (WGS), methylation profiling, and RNA sequencing (RNA-seq), we analyzed paired germline, NATs, and tumor samples from 43 individuals with BC in Hong Kong (HK). We found that single-nucleotide variants (SNVs) were common in NATs, with one-third of NAT samples exhibiting SNVs in driver genes, many of which were present in paired tumor samples. The most frequently mutated genes in both tumor and NAT samples were PIK3CA, TP53, GATA3, and AKT1. In contrast, large-scale aberrations such as somatic CN alterations (SCNAs) and structural variants (SVs) were rarely detected in NAT samples. We generated phylogenetic trees to investigate the evolutionary history of paired NAT and tumor samples. They could be categorized into tumor only, shared, and multiple-tree groups, the last of which is concordant with non-genetic field cancerization. These groups exhibited distinct genomic and epigenomic characteristics in both NAT and tumor samples. Specifically, NAT samples in the shared-tree group showed higher number of mutations, while NAT samples belonging to the multiple-tree group showed a less inflammatory tumor microenvironment (TME), characterized by a higher proportion of regulatory T cells (Tregs) and lower presence of CD14 cell populations. In summary, our findings highlight the diverse evolutionary history in BC NAT/tumor pairs and the impact of field cancerization and TME in shaping the genomic evolutionary history of tumors.
Keywords: Chinese; breast cancer; cancer genomics; clonal evolution; normal tissues adjacent to the tumor; omics analyses; whole-genome sequencing.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests Authors have no competing interests to disclose.
Figures

References
-
- Ribelles N., Perez-Villa L., Jerez J.M., Pajares B., Vicioso L., Jimenez B., de Luque V., Franco L., Gallego E., Marquez A., et al. Pattern of recurrence of early breast cancer is different according to intrinsic subtype and proliferation index. Breast Cancer Res. 2013;15 doi: 10.1186/bcr3559. - DOI - PMC - PubMed
-
- Gadaleta E., Fourgoux P., Pirró S., Thorn G.J., Nelan R., Ironside A., Rajeeve V., Cutillas P.R., Lobley A.E., Wang J., et al. Characterization of four subtypes in morphologically normal tissue excised proximal and distal to breast cancer. NPJ Breast Cancer. 2020;6:38. doi: 10.1038/s41523-020-00182-9. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
