

Molecular mechanism of GIRK2 channel gating modulated by cholesteryl hemisuccinate
- PMID: 39493862
- PMCID: PMC11527606
- DOI: 10.3389/fphys.2024.1486362
Molecular mechanism of GIRK2 channel gating modulated by cholesteryl hemisuccinate
Abstract
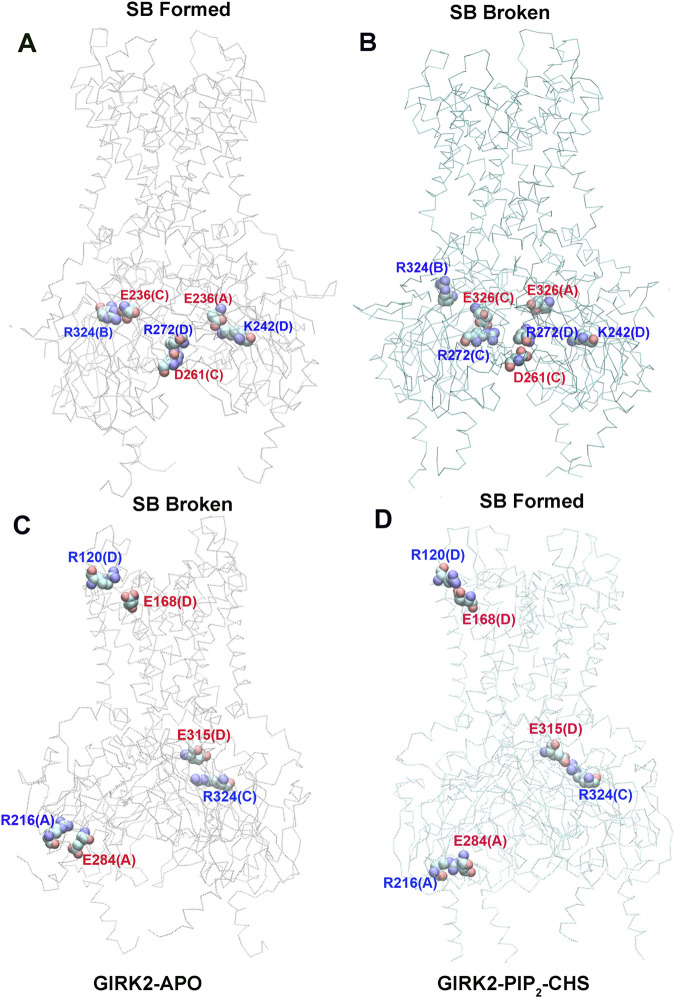
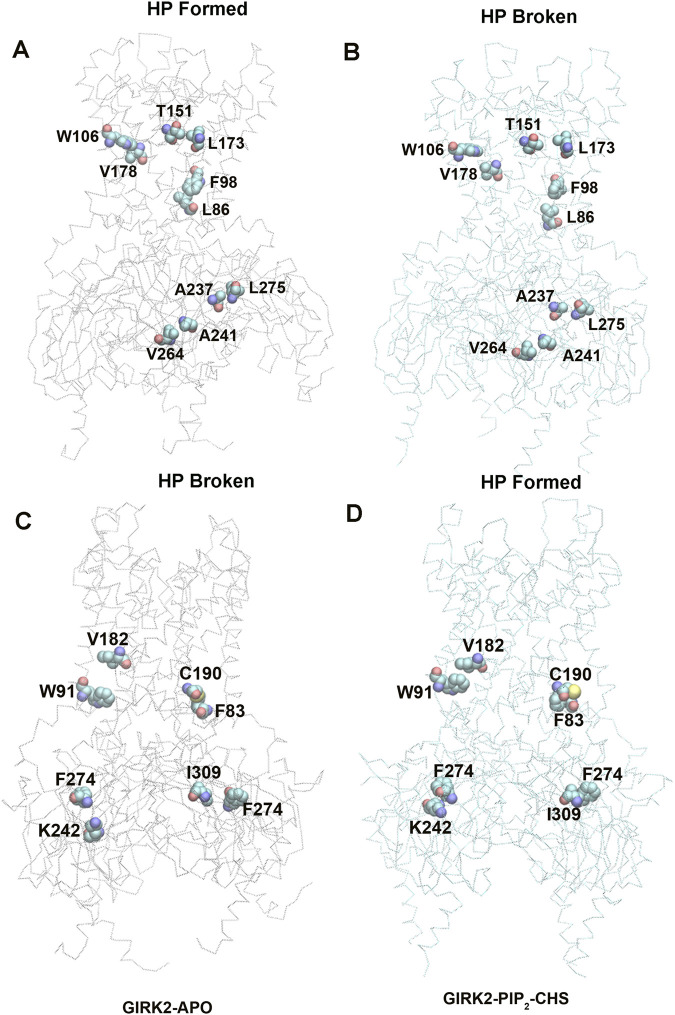
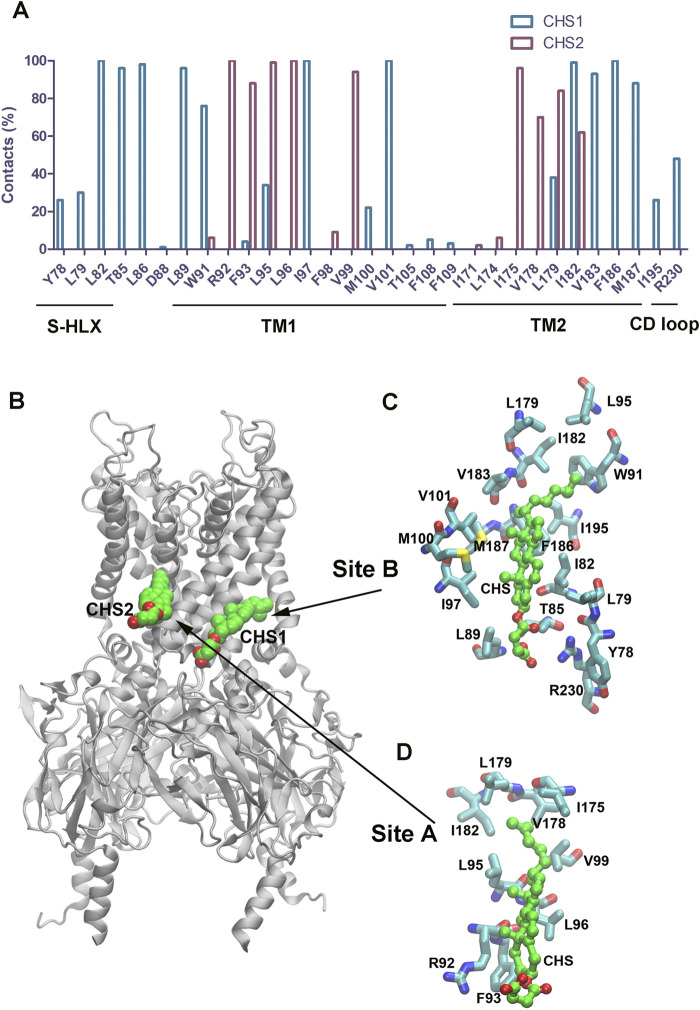
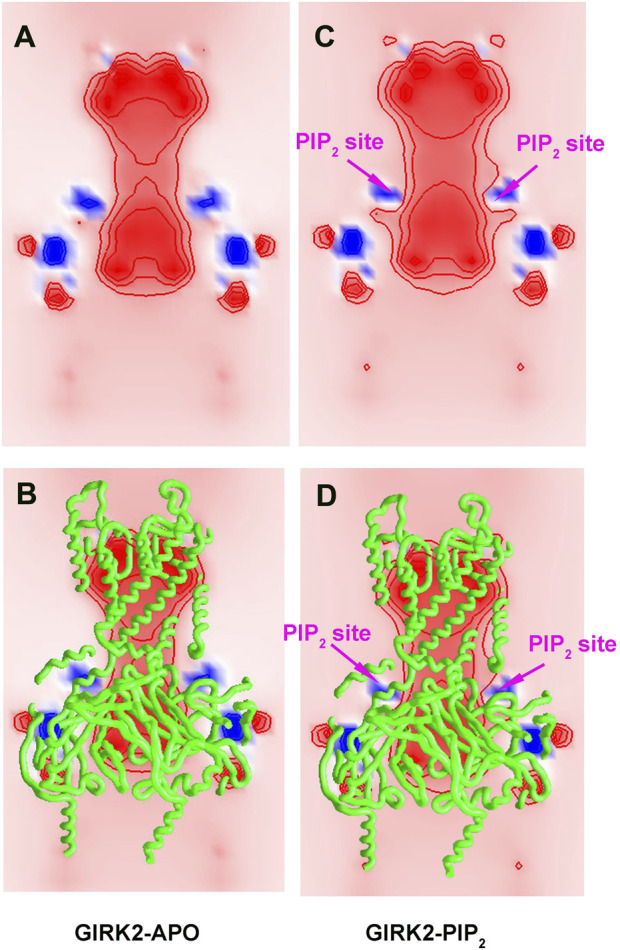
Cholesterol, an essential lipid of cell membranes, regulates G protein-gated inwardly rectifying potassium (GIRK) channel activity. Previous studies have shown that cholesterol activates GIRK2 homotetrameric channels, which are expressed in dopaminergic neurons of the brain. Deletion of GIRK2 channels affects both GIRK2 homo- and heterotetrames and can lead to abnormal neuronal excitability, including conditions such as epilepsy and addiction. A 3.5 Å cryo-EM structure of GIRK2 in complex with CHS (cholesteryl hemisuccinate) and PIP2 (phosphatidylinositol 4,5-bisphosphate) has been solved. This structure provides the opportunity to study GIRK2 channel gating dynamics regulated by cholesterol using gating molecular dynamics (GMD) simulations. In the present study, we conducted microsecond-long GMD simulations on the GIRK2 channel in its APO, PIP2, and PIP2/CHS bound states, followed by systematic analysis to gain molecular insights into how CHS modulates GIRK2 channel gating. We found that CHS binding facilitates GIRK2 channel opening, with 43 K+ ion permeation events observed, compared to 0 and 2 K+ ion permeation events for GIRK2-APO and GIRK2/PIP2, respectively. Binding of CHS to the GIRK2 channel enhances PIP2 and channel interactions, which is consistent with previous experimental results. The negatively charged PIP2 alters the internal electrostatic potential field in the channel and lowers the negative free energy barrier for K+ ion permeation.
Keywords: GIRK channel; MD simulations; cholesterol; conformational changes; electrostatic potential; interaction network analysis; ion channel activation; protein-ligand interactions.
Copyright © 2024 Cui, Lu, Ma and Logothetis.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Figures












References
-
- Amadei A., Ceruso M. A., Di Nola A. (1999). On the convergence of the conformational coordinates basis set obtained by the essential dynamics analysis of proteins' molecular dynamics simulations. Proteins 36 (4), 419–424. 10.1002/(sici)1097-0134(19990901)36:4<419::aid-prot5>3.0.co;2-u - DOI - PubMed
-
- Breneman C. M., Wiberg K. B. (1990). Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 11, 361–373. 10.1002/jcc.540110311 - DOI
Grants and funding
LinkOut - more resources
Full Text Sources