

The Neuroprotective Mechanisms of PPAR-γ: Inhibition of Microglia-Mediated Neuroinflammation and Oxidative Stress in a Neonatal Mouse Model of Hypoxic-Ischemic White Matter Injury
- PMID: 39496476
- PMCID: PMC11534457
- DOI: 10.1111/cns.70081
The Neuroprotective Mechanisms of PPAR-γ: Inhibition of Microglia-Mediated Neuroinflammation and Oxidative Stress in a Neonatal Mouse Model of Hypoxic-Ischemic White Matter Injury
Abstract
Background: Neuroinflammation and oxidative stress, mediated by microglial activation, hinder the development of oligodendrocytes (OLs) and delay myelination in preterm infants, leading to white matter injury (WMI) and long-term neurodevelopmental sequelae. Peroxisome proliferator-activated receptor gamma (PPAR-γ) has been reported to inhibit inflammation and oxidative stress via modulating microglial polarization in various central nervous system diseases. However, the relationship between PPAR-γ and microglial polarization in neonatal WMI is not well understood. Therefore, this study aimed to elucidate the role and mechanisms of PPAR-γ in preterm infants affected by WMI.
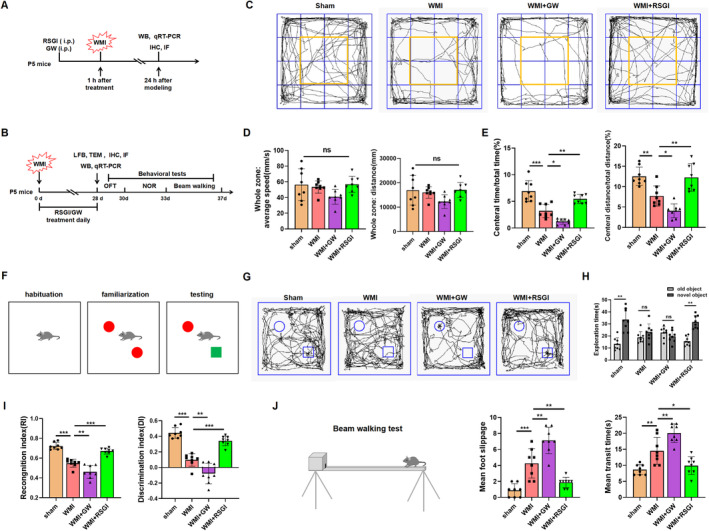
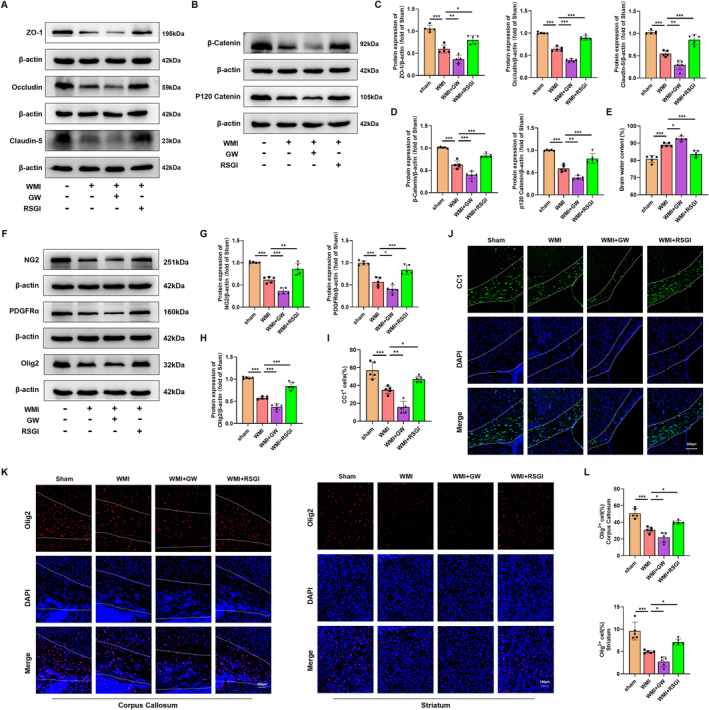
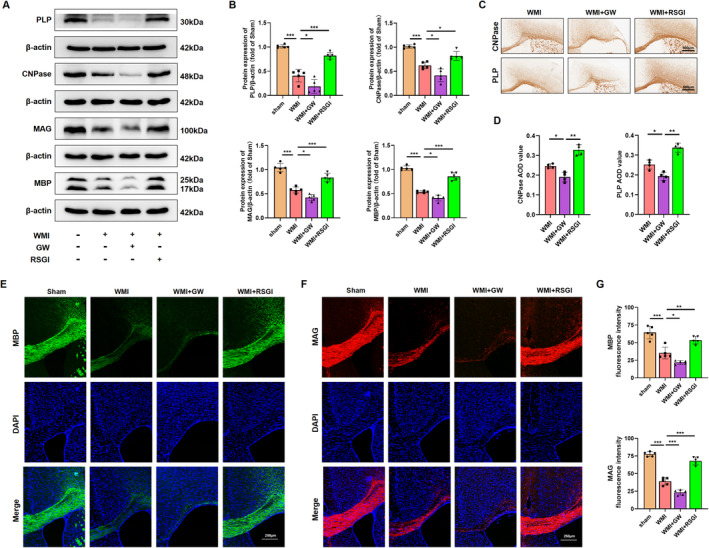
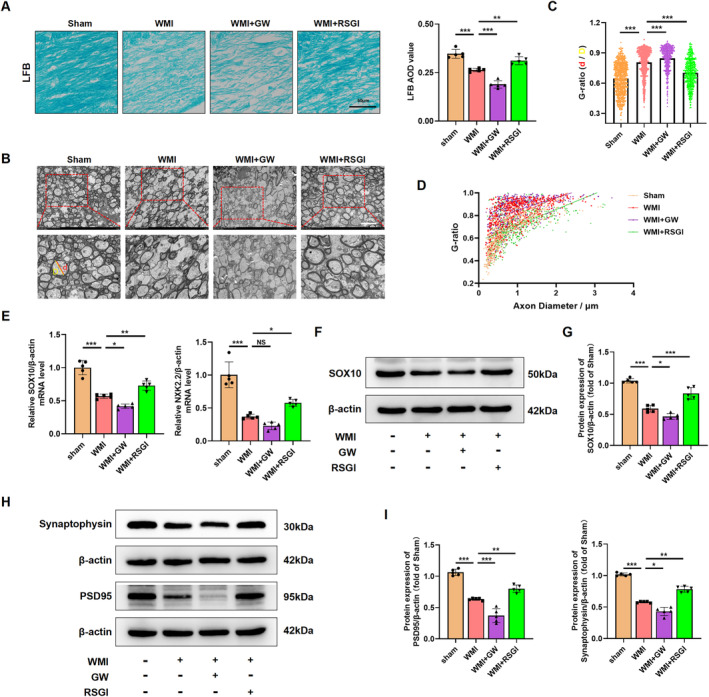
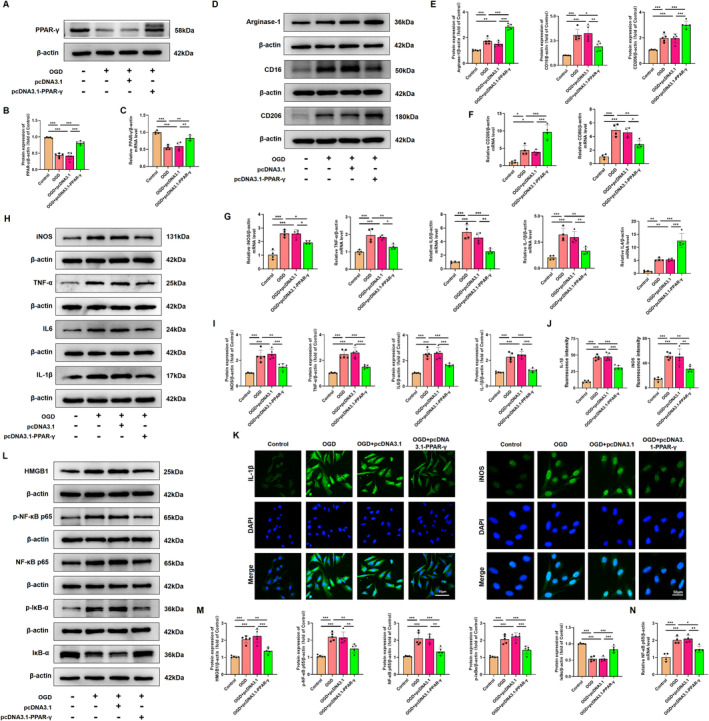
Methods: In this study, an in vivo hypoxia-ischemia (HI) induced brain WMI neonatal mouse model was established. The mice were administered intraperitoneally with either RSGI or GW9662 to activate or inhibit PPAR-γ, respectively. Additionally, an in vitro oxygen-glucose deprivation (OGD) cell model was established and pretreated with pcDNA 3.1-PPAR-γ or si-PPAR-γ to overexpress or silence PPAR-γ, respectively. The neuroprotective effects of PPAR-γ were investigated in vivo. Firstly, open field test, novel object recognization test, and beam-walking test were employed to assess the effects of PPAR-γ on neurobehavioral recovery. Furthermore, assessment of OLs loss and OL-maturation disorder, the number of myelinated axons, myelin thickness, synaptic deficit, activation of microglia and astrocyte, and blood-brain barrier (BBB) were used to evaluate the effects of PPAR-γ on pathological repair. The mechanisms of PPAR-γ were explored both in vivo and in vitro. Assessment of microglia polarization, inflammatory mediators, reactive oxygen species (ROS), MDA, and antioxidant enzymes was used to evaluate the anti-inflammatory and antioxidative effects of PPAR-γ activation. An assessment of HMGB1/NF-κB and NRF2/KEAP1 signaling pathway was conducted to clarify the mechanisms by which PPAR-γ influences HI-induced WMI in neonatal mice.
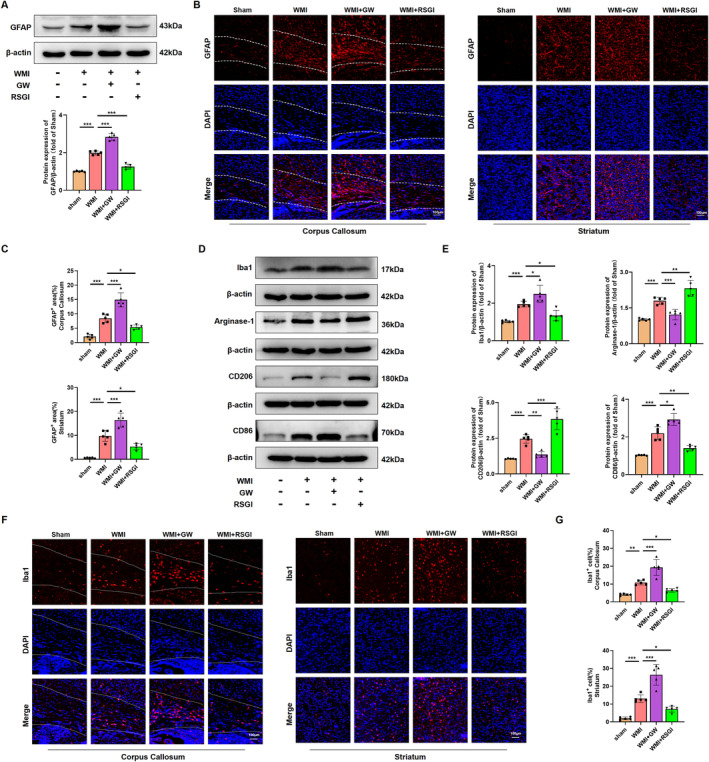
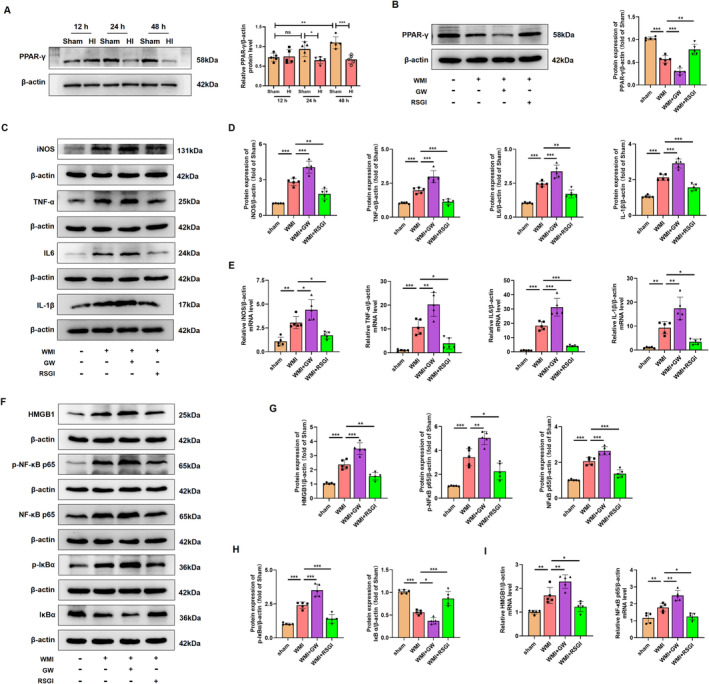
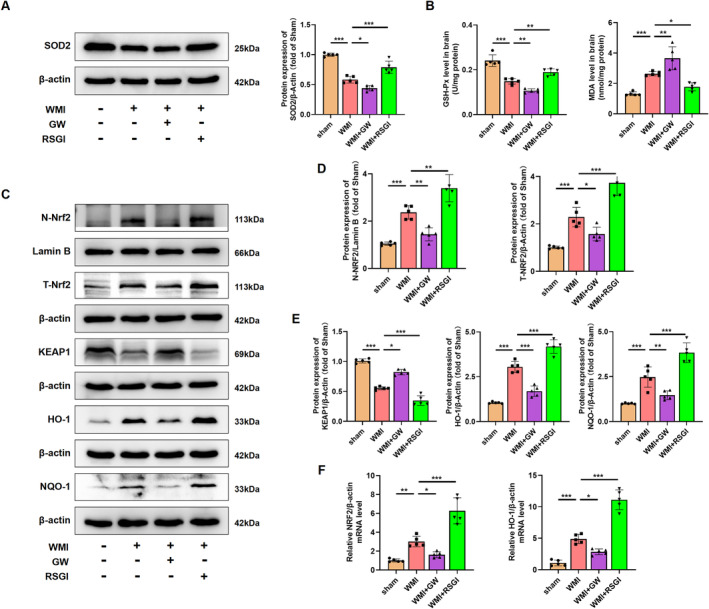
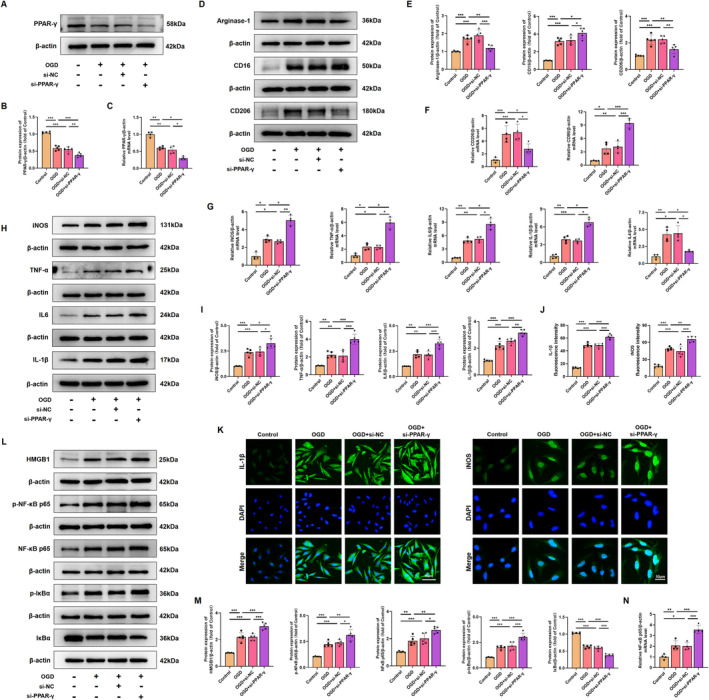
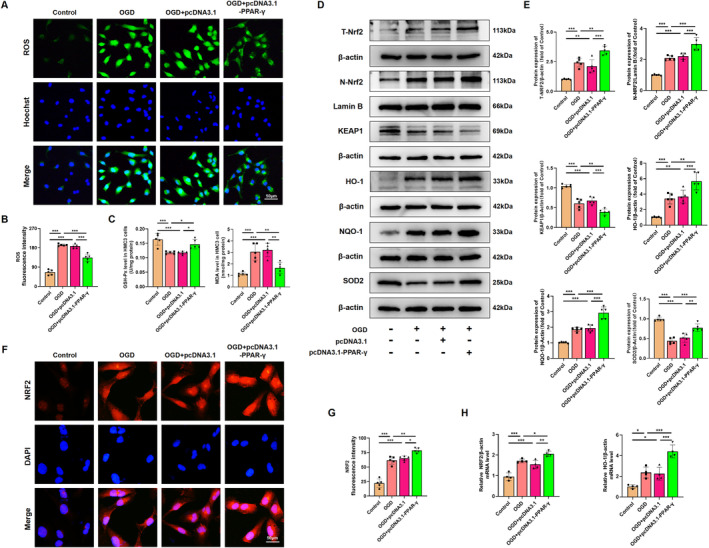
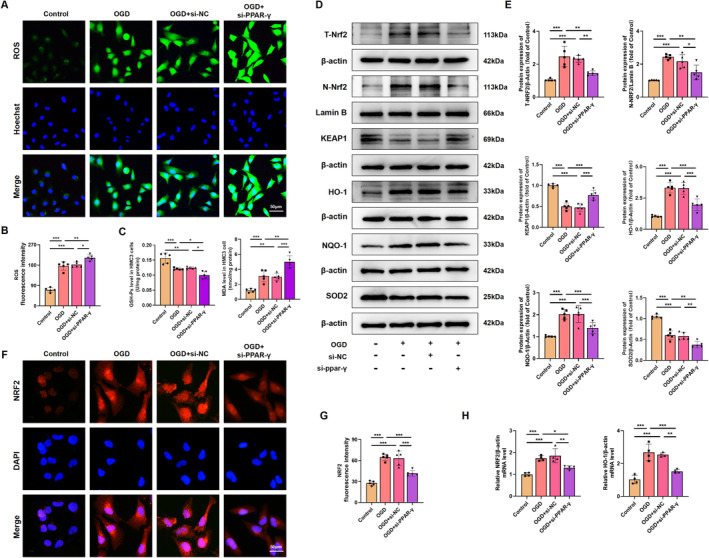
Results: Activation of PPAR-γ using RSGI significantly mitigated BBB disruption, promoted M2 polarization of microglia, inhibited activation of microglia and astrocytes, promoted OLs development, and enhanced myelination in HI-induced WMI. Conversely, inhibition of PPAR-γ using GW9662 further exacerbated the pathologic hallmark of WMI. Neurobehavioral tests revealed that neurological deficits were ameliorated by RSGI, while further aggravated by GW91662. In addition, activation of PPAR-γ significantly alleviated neuroinflammation and oxidative stress by suppressing HMGB1/NF-κB signaling pathway and activating NRF2 signaling pathway both in vivo and in vitro. Conversely, inhibition of PPAR-γ further exacerbated HI or OGD-induced neuroinflammation, oxidative stress via modulation of the same signaling pathway.
Conclusions: Our findings suggest that PPAR-γ regulates microglial activation/polarization as well as subsequent neuroinflammation/oxidative stress via the HMGB1/NF-κB and NRF2/KEAP1 signaling pathway, thereby contributing to neuroprotection and amelioration of HI-induced WMI in neonatal mice.
Keywords: PPAR‐γ; microglia; neuroinflammation; oxidative stress; white matter injury.
© 2024 The Author(s). CNS Neuroscience & Therapeutics published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Benavente‐Fernandez I., Siddiqi A., and Miller S. P., “Socioeconomic Status and Brain Injury in Children Born Preterm: Modifying Neurodevelopmental Outcome,” Pediatric Research 87, no. 2 (2020): 391–398. - PubMed
-
- Raju T. N. K., Buist A. S., Blaisdell C. J., Moxey‐Mims M., and Saigal S., “Adults Born Preterm: A Review of General Health and System‐Specific Outcomes,” Acta Paediatrica 106, no. 9 (2017): 1409–1437. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
