

Allostery in homodimeric SARS-CoV-2 main protease
- PMID: 39496839
- PMCID: PMC11535432
- DOI: 10.1038/s42003-024-07138-w
Allostery in homodimeric SARS-CoV-2 main protease
Abstract
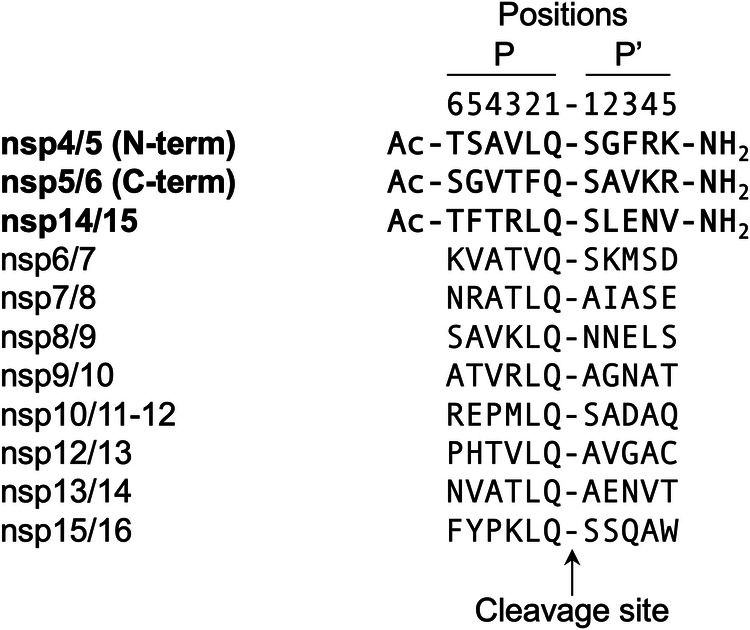
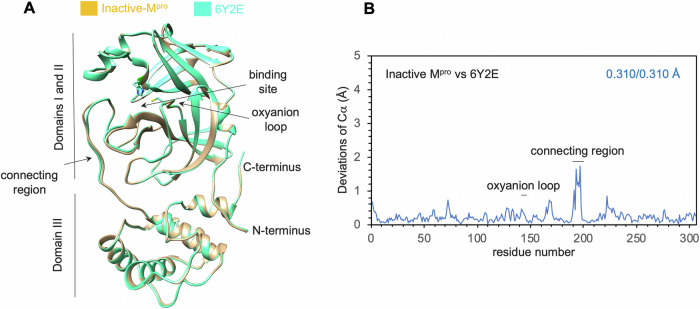
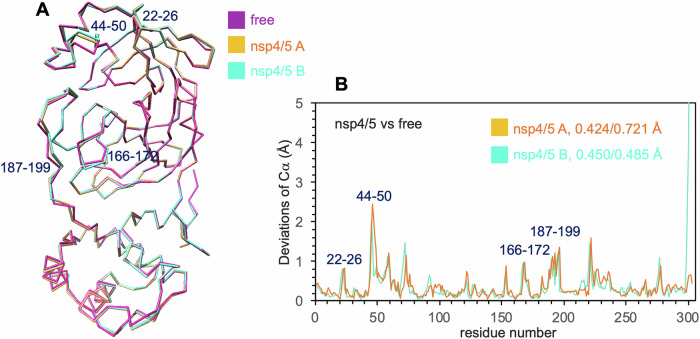
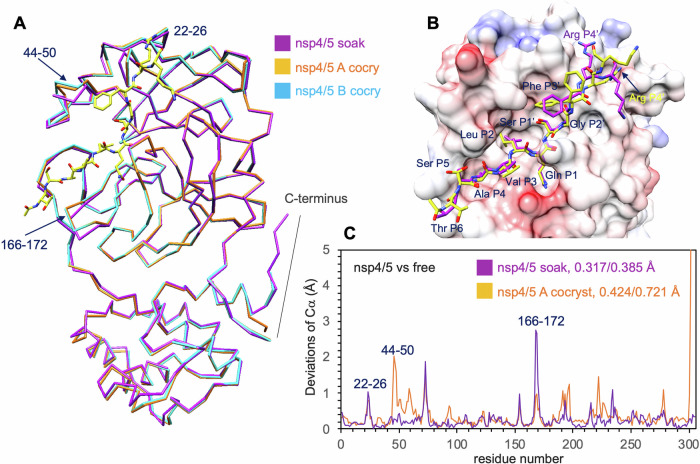
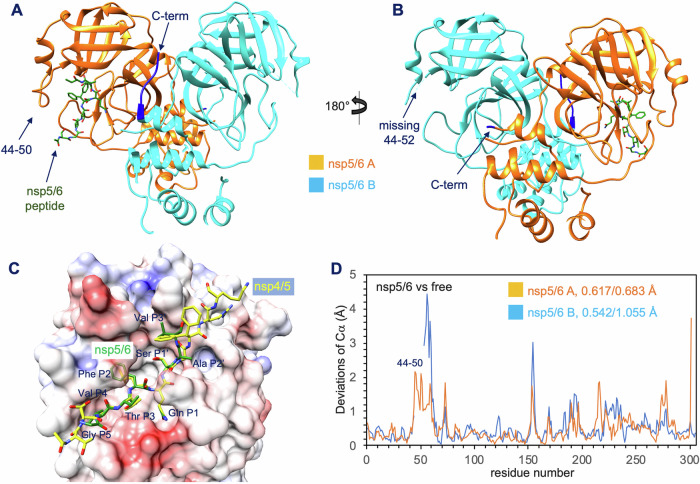
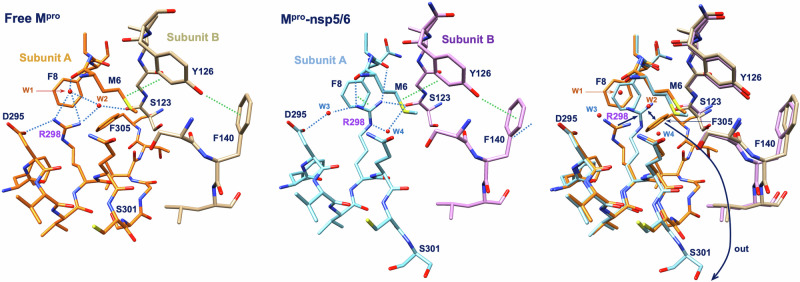
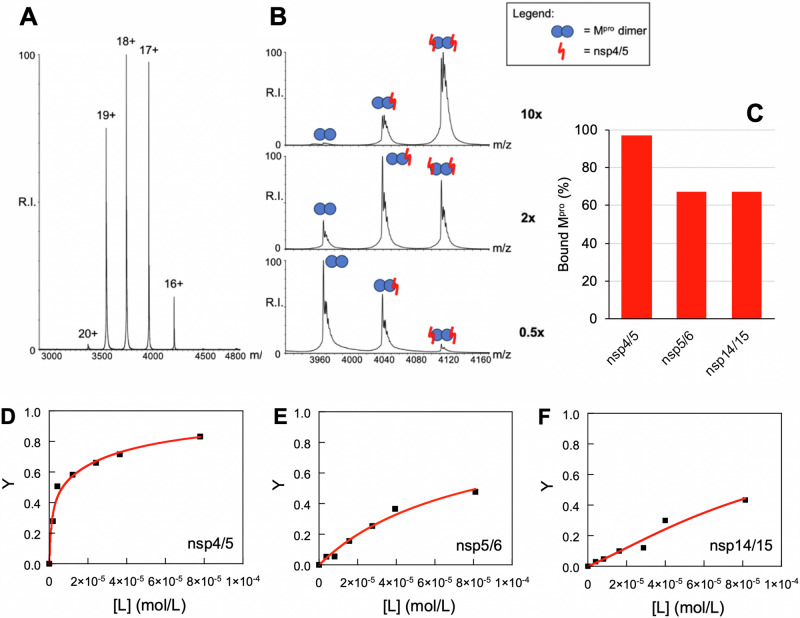
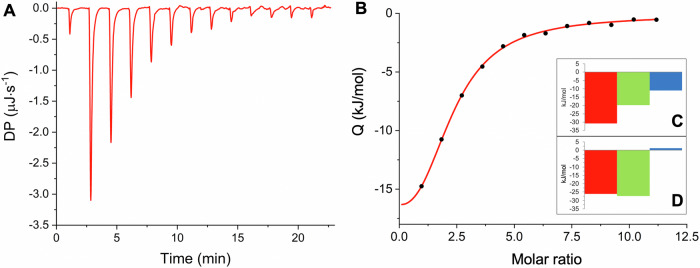
Many enzymes work as homodimers with two distant catalytic sites, but the reason for this choice is often not clear. For the main protease Mpro of SARS-CoV-2, dimerization is essential for function and plays a regulatory role during the coronaviral replication process. Here, to analyze a possible allosteric mechanism, we use X-ray crystallography, native mass spectrometry, isothermal titration calorimetry, and activity assays to study the interaction of Mpro with three peptide substrates. Crystal structures show how the plasticity of Mpro is exploited to face differences in the sequences of the natural substrates. Importantly, unlike in the free form, the Mpro dimer in complex with these peptides is asymmetric and the structures of the substrates nsp5/6 and nsp14/15 bound to a single subunit show allosteric communications between active sites. We identified arginines 4 and 298 as key elements in the transition from symmetric to asymmetric dimers. Kinetic data allowed the identification of positive cooperativity based on the increase in the processing efficiency (kinetic allostery) and not on the better binding of the substrates (thermodynamic allostery). At the physiological level, this allosteric behavior may be justified by the need to regulate the processing of viral polyproteins in time and space.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Kim, T. H. et al. The role of dimer asymmetry and protomer dynamics in enzyme catalysis. Science355, eaag2355 (2017). - PubMed
MeSH terms
Substances
Grants and funding
- Progetto di ricerca sul Covid-19 N° 55812/Fondazione Cassa di Risparmio di Padova e Rovigo (Foundation Cariparo)
- PRIN 2017, code 2017BZEREZ_003/Ministero dell'Istruzione, dell'Università e della Ricerca (Ministry of Education, University and Research)
- PRIN 2022, code 2022Y5C7KL/Ministero dell'Istruzione, dell'Università e della Ricerca (Ministry of Education, University and Research)
- P-DiSC, BIRD2023-UNIPD/Università degli Studi di Padova (University of Padova)
LinkOut - more resources
Full Text Sources
Miscellaneous
