

Blockade of 11β-hydroxysteroid dehydrogenase type 1 ameliorates metabolic dysfunction-associated steatotic liver disease and fibrosis
- PMID: 39498052
- PMCID: PMC11534184
- DOI: 10.1016/j.heliyon.2024.e39534
Blockade of 11β-hydroxysteroid dehydrogenase type 1 ameliorates metabolic dysfunction-associated steatotic liver disease and fibrosis
Abstract
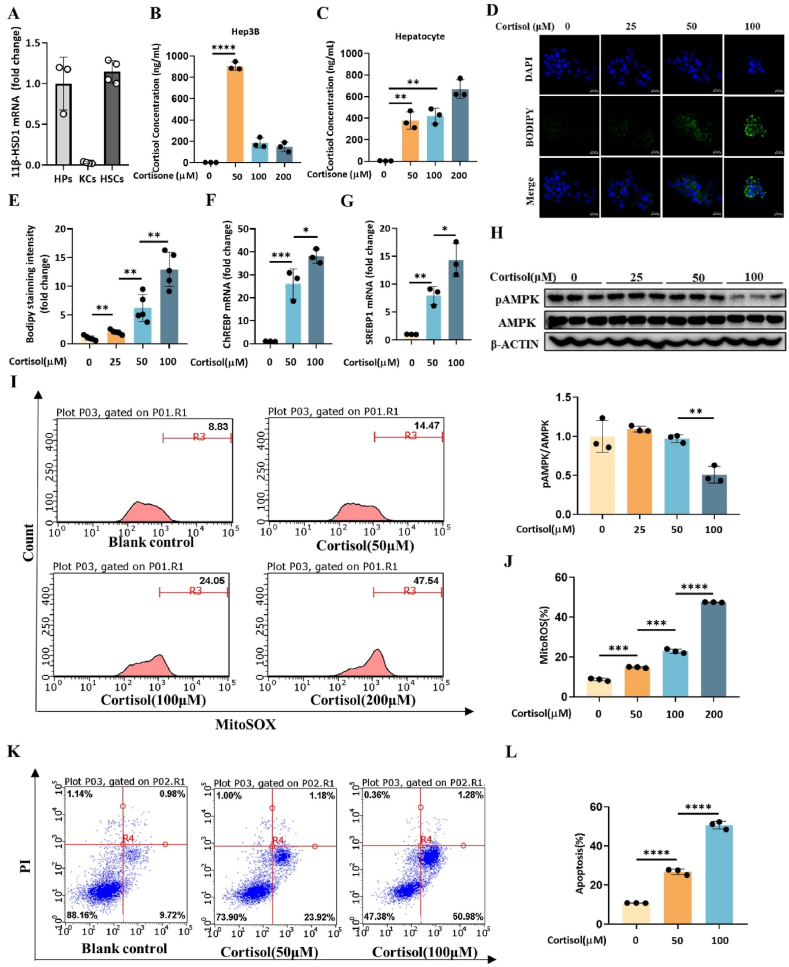
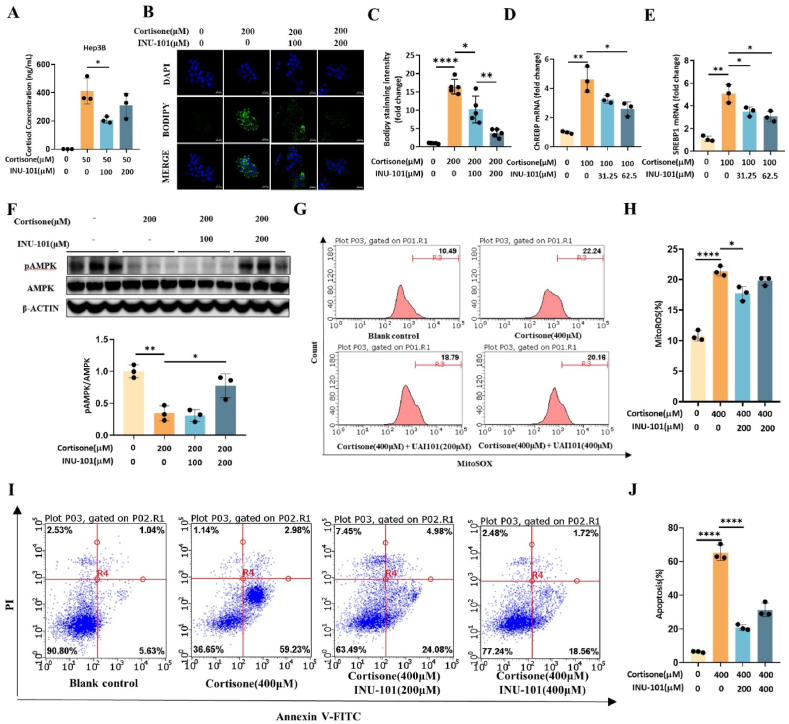
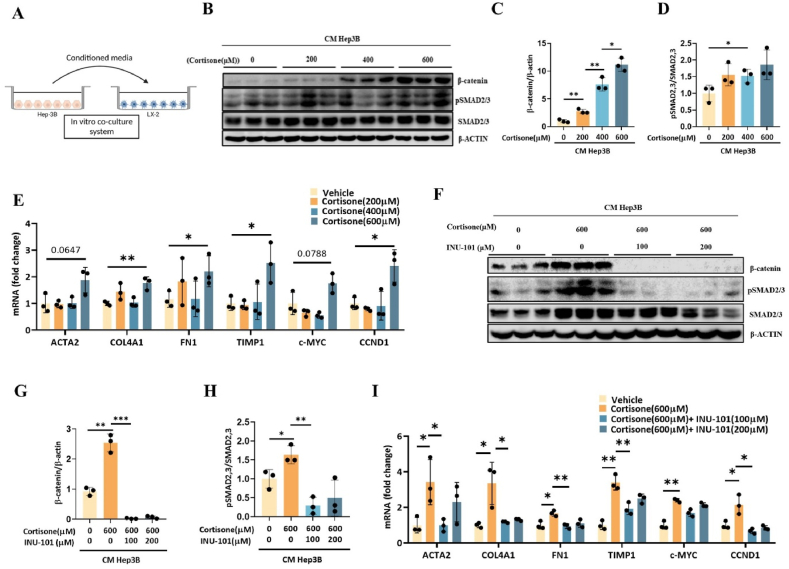
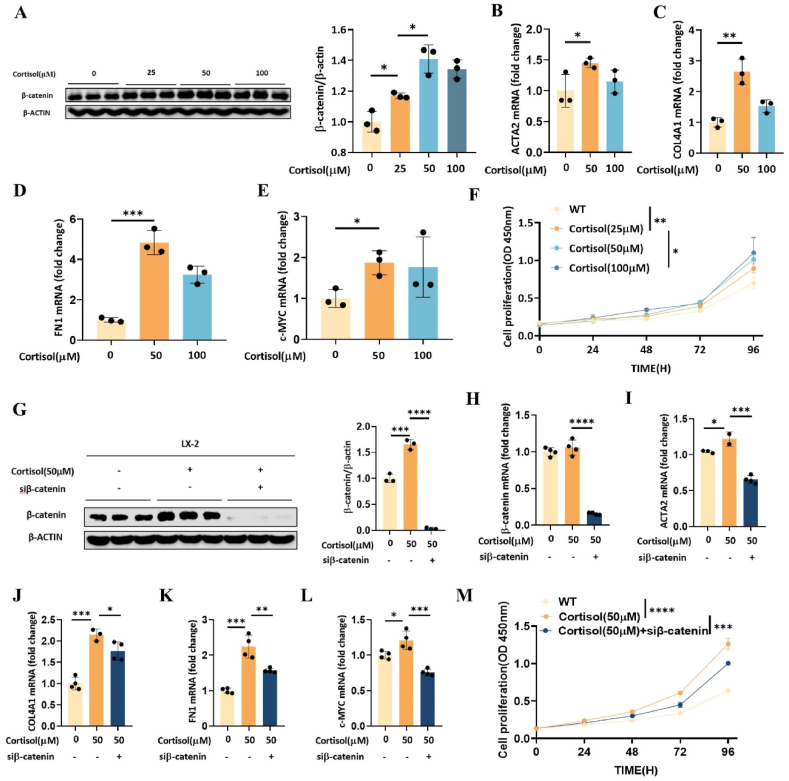
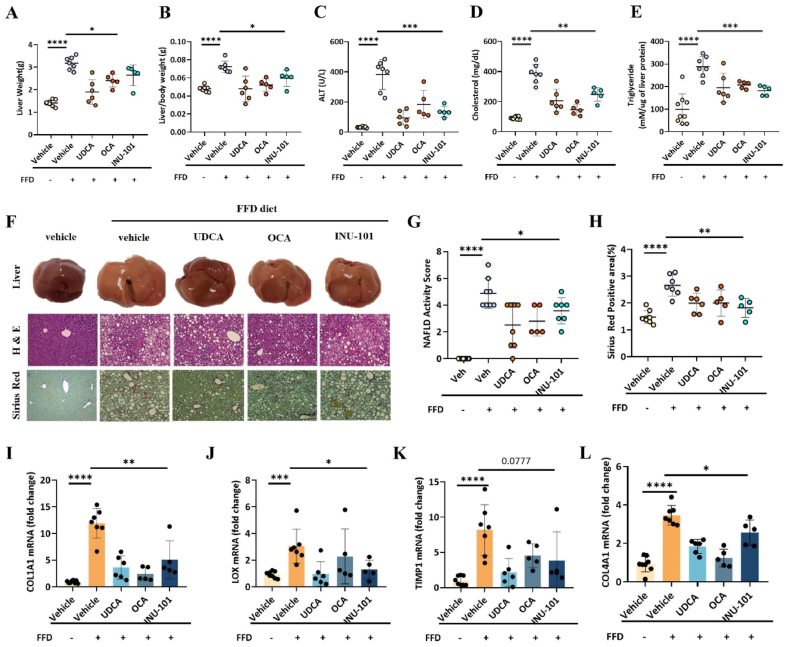
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) is a key enzyme involved in the conversion of cortisone to active cortisol in the liver. Elevated cortisol levels can trigger oxidative stress, inflammation, and hepatocyte damage, highlighting the importance of 11β-HSD1 inhibition as a potential therapeutic approach. This study aimed to explore the effects of INU-101, an inhibitor of 11β-HSD1, on the development of metabolic dysfunction-associated steatotic liver disease (MASLD) and fibrosis. Our findings demonstrated that INU-101 effectively mitigated cortisol-induced lipid accumulation, reactive oxygen species generation, and hepatocyte apoptosis. Furthermore, 11β-HSD1 inhibition suppressed hepatic stellate cell activation by modulating β-catenin and phosphorylated SMAD2/3. INU-101 administration significantly reduced hepatic lipid accumulation and liver fibrosis in mice fed fast-food diet. This study suggests that INU-101 holds promise as a clinical candidate for treating MASLD and fibrosis, offering potential therapeutic benefits by targeting the intricate processes involving 11β-HSD1 and cortisol regulation in the liver.
Keywords: 11β-HSD1; Cortisol; Cortisone; Lipid accumulation; Liver fibrosis.
© 2024 Published by Elsevier Ltd.
Conflict of interest statement
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Jun Sung Jang reports equipment, drugs, or supplies was provided by Ahn-Gook Phamaceutical Co., Ltd. Soo Im Chang reports equipment, drugs, or supplies was provided by Ahn-Gook Phamaceutical Co., Ltd. Jun Sung Jang reports a relationship with Ahn-Gook Phamaceutical Co., Ltd. that includes: employment. Soo Im Chang reports a relationship with Ahn-Gook Phamaceutical Co., Ltd. that includes: employment. INU-101 is under intellectual property rights owned by Ahn-Kook Pharmaceutical Co., Ltd., Korea, and we conducted experiments with the provision of INU-101, UDCA, and OCA. If there are other authors, they declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
-
- Duell P.B., Welty F.K., Miller M., Chait A., Hammond G., Ahmad Z., Cohen D.E., Horton J.D., Pressman G.S., Toth P.P., T. American Heart Association Council on Arteriosclerosis, Vascular B., Council on H., D. Council on the Kidney in Cardiovascular, Council on L., Cardiometabolic H., Council D. On peripheral Vascular.Nonalcoholic fatty liver disease and cardiovascular risk: a scientific statement from the American heart association. Arterioscler. Thromb. Vasc. Biol. 2022;42:e168–e185. doi: 10.1161/ATV.0000000000000153. - DOI - PubMed
-
- Harrison S.A., Bedossa P., Guy C.D., Schattenberg J.M., Loomba R., Taub R., Labriola D., Moussa S.E., Neff G.W., Rinella M.E., Anstee Q.M., Abdelmalek M.F., Younossi Z., Baum S.J., Francque S., Charlton M.R., Newsome P.N., Lanthier N., Schiefke I., Mangia A., Pericas J.M., Patil R., Sanyal A.J., Noureddin M., Bansal M.B., Alkhouri N., Castera L., Rudraraju M., Ratziu V., Investigators M.-N. A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. N. Engl. J. Med. 2024;390:497–509. doi: 10.1056/NEJMoa2309000. - DOI - PubMed
LinkOut - more resources
Full Text Sources
