

Development of novel CDK9 and CYP3A4 inhibitors for cancer therapy through field and computational approaches
- PMID: 39498375
- PMCID: PMC11532072
- DOI: 10.3389/fchem.2024.1473398
Development of novel CDK9 and CYP3A4 inhibitors for cancer therapy through field and computational approaches
Abstract
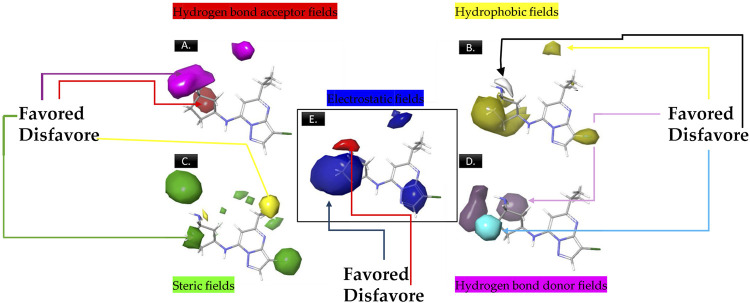
Cyclin-dependent kinase 9 (CDK9) and cytochrome P450 3A4 (CYP3A4) have emerged as promising targets in the development of anticancer drugs, presenting a consistent challenge in the quest for potent inhibitors. CDK9 inhibitors can selectively target fast-growing cancer cells by disrupting transcription elongation, which in turn hinders the production of proteins essential for cell cycle progression and survivaŚ. Understanding how CYP3A4 metabolizes specific chemotherapy drugs allows for personalized treatment plans, optimizing drug dosages according to a patient's metabolic profile. Since many cancer patients undergo combination therapies, and CYP3A4 is vital in drug metabolism, its inhibition or induction by one drug can alter the plasma levels of others, potentially leading to treatment failure or increased toxicity. Therefore, managing CYP3A4 activity is critical for effective cancer treatment. Employing a range of computational methodologies, this study systematically investigated the binding mechanisms of pyrimidine derivatives against CDK9 and CYP3A4. The field-based model demonstrated high R 2 values (0.99), with Q2 (0.66), demonstrating its ability to predict in silico inhibitory activity against the target of this study. The screening process followed in this work led to the discovery of powerful new inhibitor compounds. Of the 15 new compounds designed, three have a high affinity with the target (ranging from -8 to -9 kcal/mol kcal/mol) and were singled out through docking filtration for more detailed investigation. As well as, a reference compound with a substantial pIC50 value of 8.4, serving as the foundation for the development of the new compounds, was included for comparative analysis. To elucidate the essential features of CDK9 and CYP3A4 inhibitor design, a comparative analysis was conducted between 3D-QSAR-generated contours and molecular docking conformations of ligands. Molecular dynamics simulations were carried out for a duration of 100 ns on selected docked complexes, specifically those involving novel compounds with CDK9 and CYP3A4 enzymes. Additionally, the binding free energy for these complexes was assessed using the MM/PBSA method, which evaluates the free energy landscape of protein-ligand interactions. The results of MM/PBSA highlighted the strength of the new compounds in enhancing interactions with the target protein, which favors the results of molecular docking and MD simulation. These insights contribute to a deeper understanding of the mechanisms underlying CDK9 and CYP3A4 inhibition, offering potential avenues for the development of innovative and effective CDK9 inhibitors.
Keywords: 3D-QSAR; CADD; CDK9; Cancer; drug design.
Copyright © 2024 Alsfouk, Faris, Cacciatore and Alnajjar.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures










Similar articles
-
Integrated computational approaches for designing potent pyrimidine-based CDK9 inhibitors: 3D-QSAR, docking, and molecular dynamics simulations.Comput Biol Chem. 2024 Feb;108:108003. doi: 10.1016/j.compbiolchem.2023.108003. Epub 2023 Dec 12. Comput Biol Chem. 2024. PMID: 38159453
-
Uncovering potential CDK9 inhibitors from natural compound databases through docking-based virtual screening and MD simulations.J Mol Model. 2024 Jul 16;30(8):267. doi: 10.1007/s00894-024-06067-z. J Mol Model. 2024. PMID: 39012568
-
Identification of novel CDK 9 inhibitors based on virtual screening, molecular dynamics simulation, and biological evaluation.Life Sci. 2020 Oct 1;258:118228. doi: 10.1016/j.lfs.2020.118228. Epub 2020 Aug 8. Life Sci. 2020. PMID: 32781071
-
In silico design of novel FAK inhibitors using integrated molecular docking, 3D-QSAR and molecular dynamics simulation studies.J Biomol Struct Dyn. 2022 Aug;40(13):5965-5982. doi: 10.1080/07391102.2021.1875880. Epub 2021 Jan 21. J Biomol Struct Dyn. 2022. PMID: 33475043
-
Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4.Curr Drug Metab. 2008 May;9(4):310-22. doi: 10.2174/138920008784220664. Curr Drug Metab. 2008. PMID: 18473749 Review.
Cited by
-
How Plant Polyhydroxy Flavonoids Can Hinder the Metabolism of Cytochrome 3A4.Biomedicines. 2025 Mar 7;13(3):655. doi: 10.3390/biomedicines13030655. Biomedicines. 2025. PMID: 40149631 Free PMC article.
References
-
- Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., et al. (2015). GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25. 10.1016/j.softx.2015.06.001 - DOI
-
- Al-Karmalawy A. A., Mousa M. H. A., Sharaky M., Mourad M. A. E., El-Dessouki A. M., Hamouda A. O., et al. (2023). Lead optimization of BIBR1591 to improve its telomerase inhibitory activity: design and synthesis of novel four chemical series with in silico, in vitro, and in vivo preclinical assessments. J. Med. Chem. 67, 492–512. 10.1021/acs.jmedchem.3c01708 - DOI - PubMed
-
- Alsfouk A. A., Alshibl H. M., Alsfouk B. A., Altwaijry N. A., Al-Abdullah E. S. (2022). Synthesis and biological evaluation of imadazo[1,2-a]pyrazines as anticancer and antiviral agents through inhibition of CDK9 and human coronavirus. Pharmaceuticals 15, 859. 10.3390/ph15070859 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous