

Tumor purity estimated from bulk DNA methylation can be used for adjusting beta values of individual samples to better reflect tumor biology
- PMID: 39498434
- PMCID: PMC11532792
- DOI: 10.1093/nargab/lqae146
Tumor purity estimated from bulk DNA methylation can be used for adjusting beta values of individual samples to better reflect tumor biology
Abstract
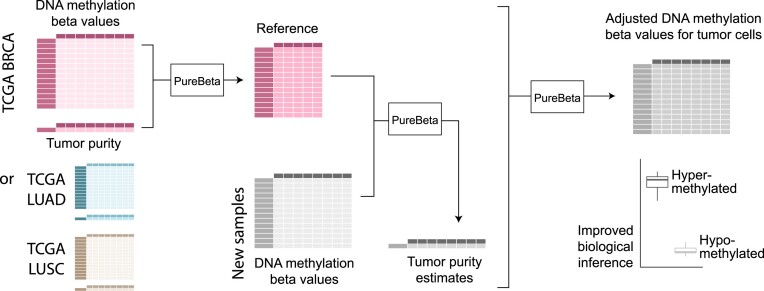
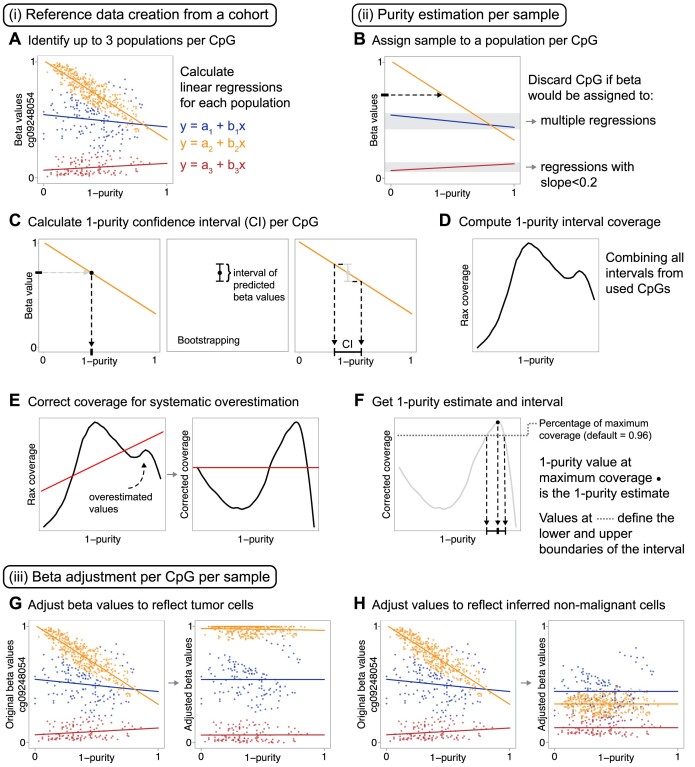
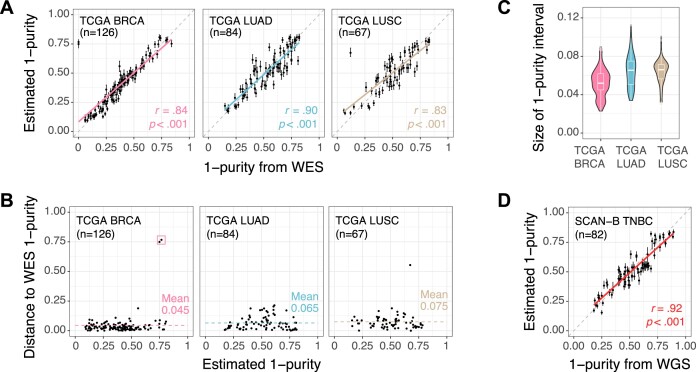
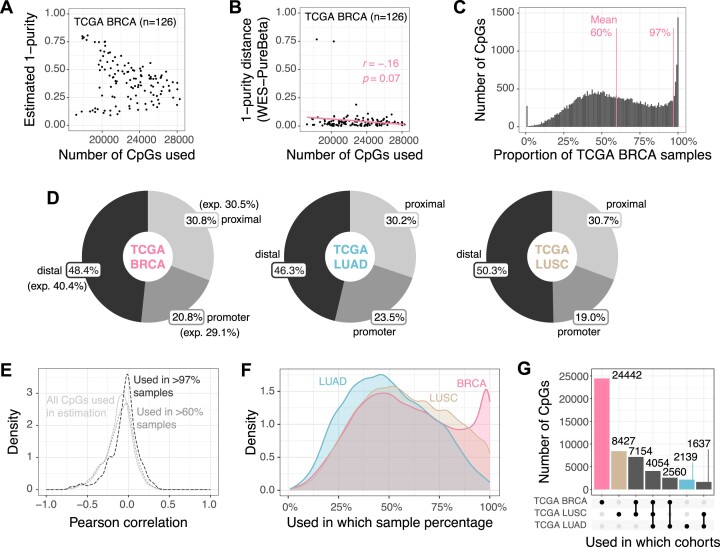
Epigenetic deregulation through altered DNA methylation is a fundamental feature of tumorigenesis, but tumor data from bulk tissue samples contain different proportions of malignant and non-malignant cells that may confound the interpretation of DNA methylation values. The adjustment of DNA methylation data based on tumor purity has been proposed to render both genome-wide and gene-specific analyses more precise, but it requires sample purity estimates. Here we present PureBeta, a single-sample statistical framework that uses genome-wide DNA methylation data to first estimate sample purity and then adjust methylation values of individual CpGs to correct for sample impurity. Purity values estimated with the algorithm have high correlation (>0.8) to reference values obtained from DNA sequencing when applied to samples from breast carcinoma, lung adenocarcinoma, and lung squamous cell carcinoma. Methylation beta values adjusted based on purity estimates have a more binary distribution that better reflects theoretical methylation states, thus facilitating improved biological inference as shown for BRCA1 in breast cancer. PureBeta is a versatile tool that can be used for different Illumina DNA methylation arrays and can be applied to individual samples of different cancer types to enhance biological interpretability of methylation data.
© The Author(s) 2024. Published by Oxford University Press on behalf of NAR Genomics and Bioinformatics.
Figures
Similar articles
-
Tumor purity adjusted beta values improve biological interpretability of high-dimensional DNA methylation data.PLoS One. 2022 Sep 9;17(9):e0265557. doi: 10.1371/journal.pone.0265557. eCollection 2022. PLoS One. 2022. PMID: 36084090 Free PMC article.
-
PESM: A novel approach of tumor purity estimation based on sample specific methylation sites.J Bioinform Comput Biol. 2020 Oct;18(5):2050027. doi: 10.1142/S0219720020500274. Epub 2020 Aug 6. J Bioinform Comput Biol. 2020. PMID: 32757807
-
RF_Purify: a novel tool for comprehensive analysis of tumor-purity in methylation array data based on random forest regression.BMC Bioinformatics. 2019 Aug 16;20(1):428. doi: 10.1186/s12859-019-3014-z. BMC Bioinformatics. 2019. PMID: 31419933 Free PMC article.
-
Tumor purity and differential methylation in cancer epigenomics.Brief Funct Genomics. 2016 Nov;15(6):408-419. doi: 10.1093/bfgp/elw016. Epub 2016 May 19. Brief Funct Genomics. 2016. PMID: 27199459 Review.
-
The DNA methylation landscape in cancer.Essays Biochem. 2019 Dec 20;63(6):797-811. doi: 10.1042/EBC20190037. Essays Biochem. 2019. PMID: 31845735 Free PMC article. Review.
Cited by
-
The DNA methylation landscape of primary triple-negative breast cancer.Nat Commun. 2025 Mar 28;16(1):3041. doi: 10.1038/s41467-025-58158-x. Nat Commun. 2025. PMID: 40155623 Free PMC article.
References
-
- Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022; 12:31–46. - PubMed
Associated data
LinkOut - more resources
Full Text Sources
Miscellaneous
