

A diet-dependent host metabolite shapes the gut microbiota to protect from autoimmunity
- PMID: 39500329
- PMCID: PMC11660937
- DOI: 10.1016/j.celrep.2024.114891
A diet-dependent host metabolite shapes the gut microbiota to protect from autoimmunity
Abstract
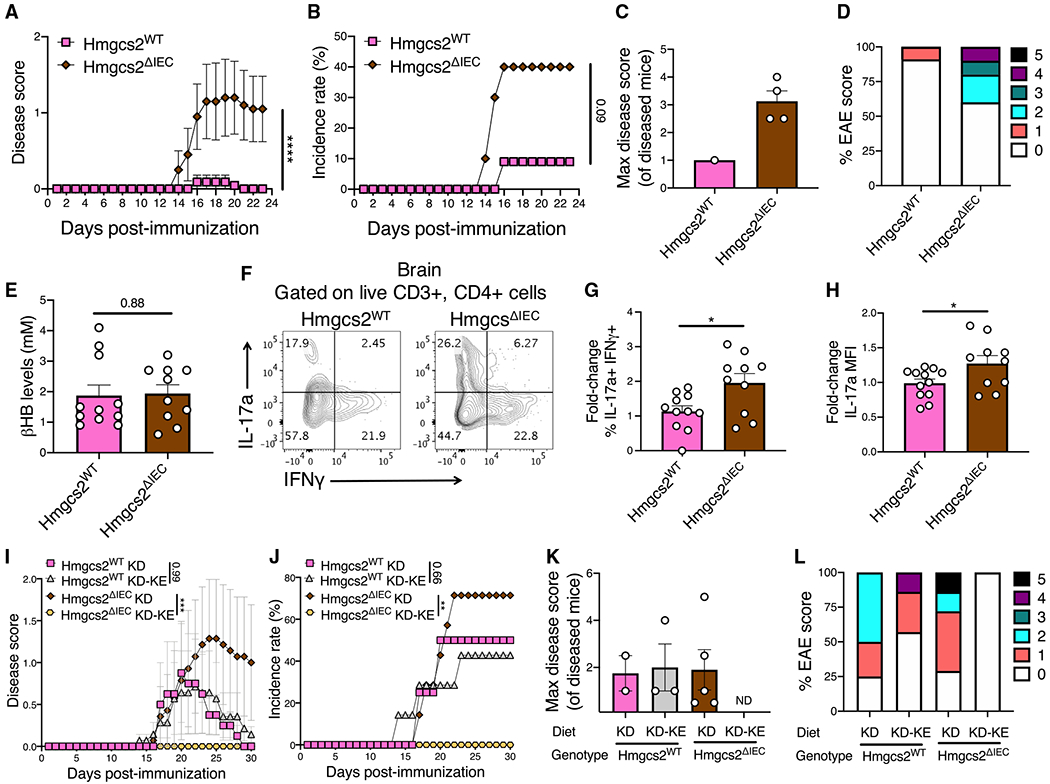
Diet can protect from autoimmune disease; however, whether diet acts via the host and/or microbiome remains unclear. Here, we use a ketogenic diet (KD) as a model to dissect these complex interactions. A KD rescued the experimental autoimmune encephalomyelitis (EAE) mouse model of multiple sclerosis in a microbiota-dependent fashion. Dietary supplementation with a single KD-dependent host metabolite (β-hydroxybutyrate [βHB]) rescued EAE, whereas transgenic mice unable to produce βHB in the intestine developed more severe disease. Transplantation of the βHB-shaped gut microbiota was protective. Lactobacillus sequence variants were associated with decreased T helper 17 cell activation in vitro. Finally, we isolated an L. murinus strain that protected from EAE, which was phenocopied by a Lactobacillus metabolite enriched by βHB supplementation, indole lactate. Thus, diet alters the immunomodulatory potential of the gut microbiota by shifting host metabolism, emphasizing the utility of taking a more integrative approach to study diet-host-microbiome interactions.
Keywords: CP: Immunology; CP: Microbiology; autoimmune disease; gut microbiome; immune activation; ketogenesis; ketogenic diet; lactobacillus; multiple sclerosis; neuroinflammation; nutrition; trytophan metabolism.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests P.J.T. is on the scientific advisory boards for Pendulum, Seed, and SNIPRbiome; there is no direct overlap between the current study and these consulting duties. P.A.C. has served as an external consultant for Pfizer, Inc., Abbott Laboratories, Janssen Research & Development, and Selah Therapeutics. J.C.N. is scientific co-founder and stockholder for BHB Therapeutics (which provided the ketone ester) and Selah Therapeutics.
Figures





Update of
-
A diet-dependent host metabolite shapes the gut microbiota to protect from autoimmunity.bioRxiv [Preprint]. 2024 May 13:2023.11.02.565382. doi: 10.1101/2023.11.02.565382. bioRxiv. 2024. Update in: Cell Rep. 2024 Nov 26;43(11):114891. doi: 10.1016/j.celrep.2024.114891. PMID: 37961209 Free PMC article. Updated. Preprint.
References
-
- Fitzpatrick JA, Melton SL, Yao CK, Gibson PR, and Halmos EP (2022). Dietary management of adults with IBD - the emerging role of dietary therapy. Nat. Rev. Gastroenterol. Hepatol 19, 652–669. - PubMed
-
- Zmora N, Suez J, and Elinav E (2019). You are what you eat: diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol 16, 35–56. - PubMed
-
- Thorburn AN, Macia L, and Mackay CR (2014). Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity 40, 833–842. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- R01 DK114034/DK/NIDDK NIH HHS/United States
- K08 AR073930/AR/NIAMS NIH HHS/United States
- R00 AI159227/AI/NIAID NIH HHS/United States
- R01 AG067333/AG/NIA NIH HHS/United States
- R01 AR074500/AR/NIAMS NIH HHS/United States
- F32 AI147456/AI/NIAID NIH HHS/United States
- R01 AG069781/AG/NIA NIH HHS/United States
- K08 HL165106/HL/NHLBI NIH HHS/United States
- R01 DK091538/DK/NIDDK NIH HHS/United States
- P30 AG044281/AG/NIA NIH HHS/United States
- R01 HL122593/HL/NHLBI NIH HHS/United States
- K99 AI159227/AI/NIAID NIH HHS/United States
- R01 AT011117/AT/NCCIH NIH HHS/United States
- T32 GM141323/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
