

Cell type mapping reveals tissue niches and interactions in subcortical multiple sclerosis lesions
- PMID: 39501036
- PMCID: PMC11614744
- DOI: 10.1038/s41593-024-01796-z
Cell type mapping reveals tissue niches and interactions in subcortical multiple sclerosis lesions
Abstract
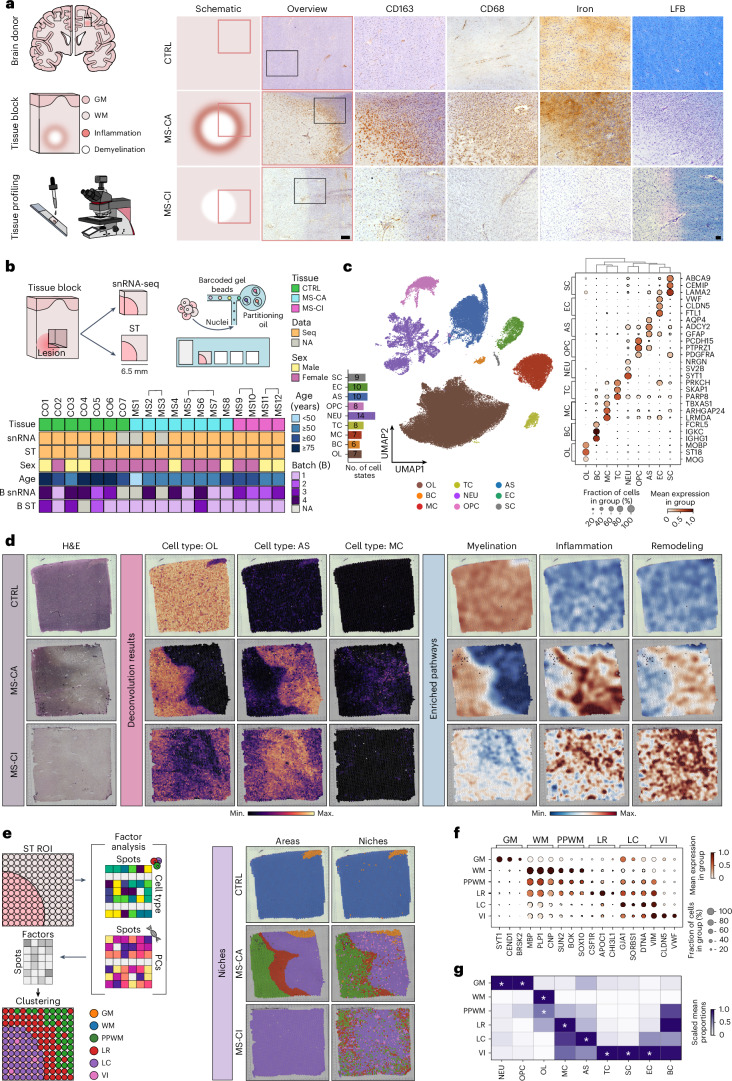
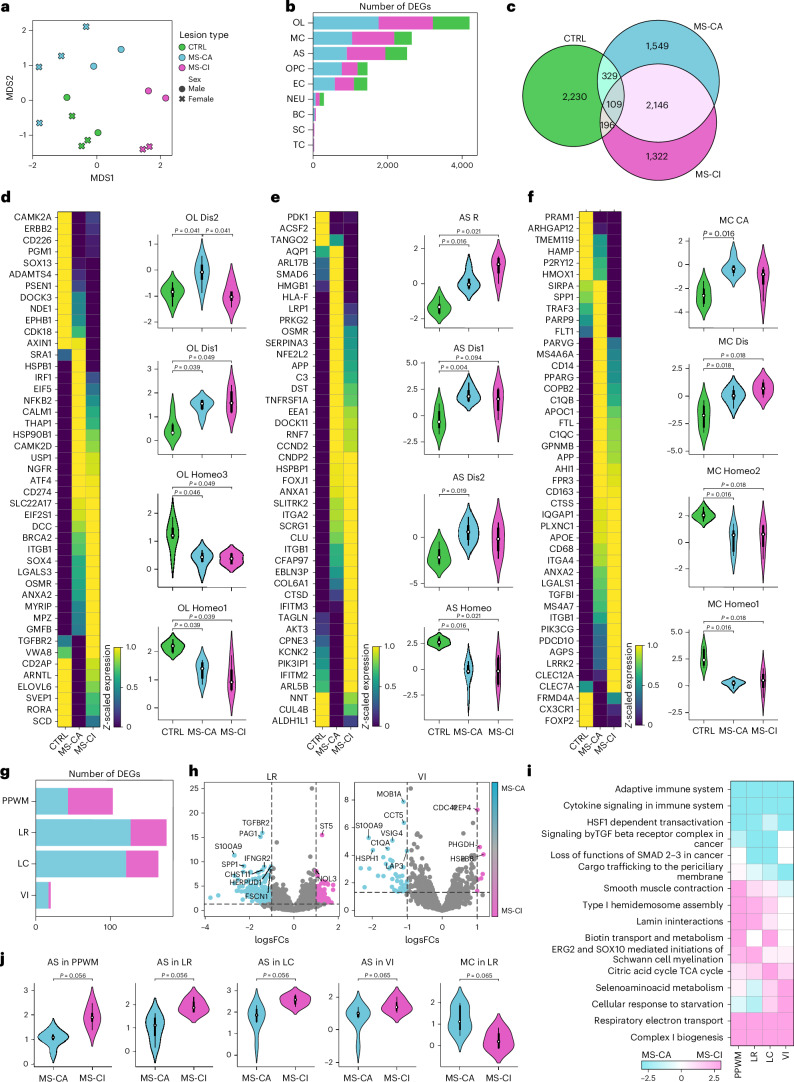
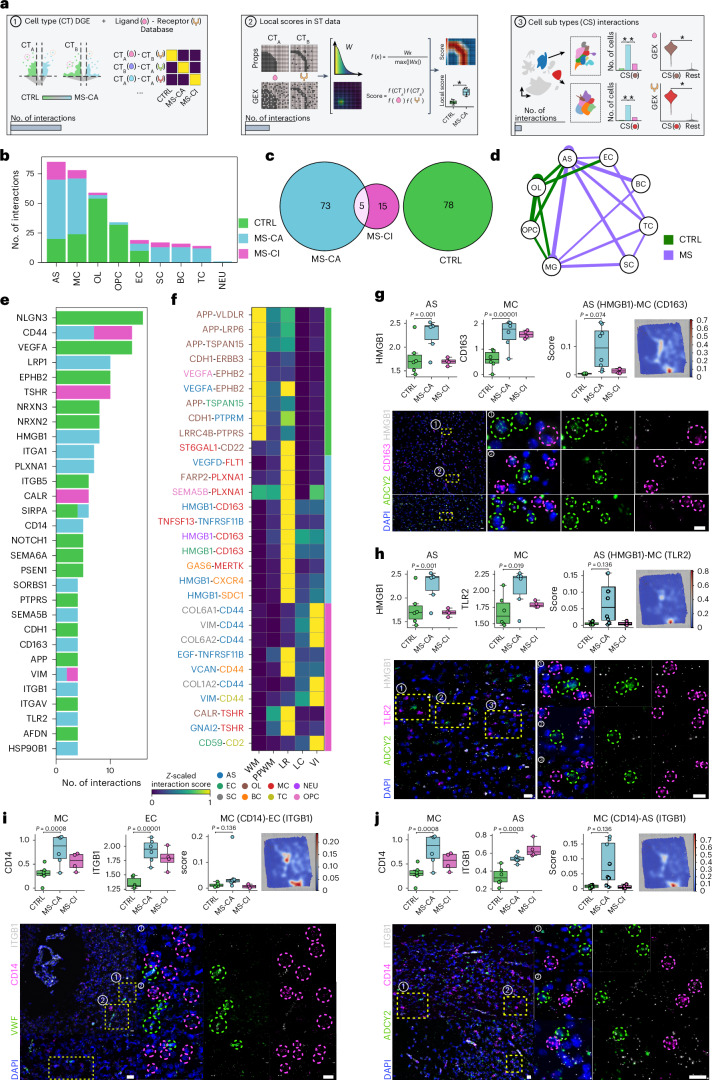
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system. Inflammation is gradually compartmentalized and restricted to specific tissue niches such as the lesion rim. However, the precise cell type composition of such niches, their interactions and changes between chronic active and inactive stages are incompletely understood. We used single-nucleus and spatial transcriptomics from subcortical MS and corresponding control tissues to map cell types and associated pathways to lesion and nonlesion areas. We identified niches such as perivascular spaces, the inflamed lesion rim or the lesion core that are associated with the glial scar and a cilia-forming astrocyte subtype. Focusing on the inflamed rim of chronic active lesions, we uncovered cell-cell communication events between myeloid, endothelial and glial cell types. Our results provide insight into the cellular composition, multicellular programs and intercellular communication in tissue niches along the conversion from a homeostatic to a dysfunctional state underlying lesion progression in MS.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: P.E. has received travel expenses from Bayer Health Care. D.S. reports funding from Cellzome, a GSK company and received honorariums from Immunai, Noetik, Alpenglow and Lunaphore. J.S.-R. reports funding from GSK, Pfizer and Sanofi and fees/honoraria from Travere Therapeutics, Stadapharm, Astex, Owkin, Moderna, Pfizer and Grunenthal. L.S. reports research support from Roche and Merck and filed a patent for the detection of antibodies against KIR4.1 in a subpopulation of patients with multiple sclerosis (WO2015166057A1). The remaining authors declare no competing interests.
Figures











References
MeSH terms
Grants and funding
- R01 NS114227/NS/NINDS NIH HHS/United States
- P1180016/Gemeinnützige Hertie-Stiftung (Hertie Foundation)
- RF1 MH132662/MH/NIMH NIH HHS/United States
- PA-2002-36405/National Multiple Sclerosis Society (National MS Society)
- R01NS114227/U.S. Department of Health & Human Services | NIH | National Institute of Neurological Disorders and Stroke (NINDS)
- RF1MH132662/U.S. Department of Health & Human Services | NIH | National Institute of Mental Health (NIMH)
- U24HG002371/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- U24 HG002371/HG/NHGRI NIH HHS/United States
- DISC0-14514/California Institute for Regenerative Medicine (CIRM)
- PA-2002-36405, RFA-2203-39300/National Multiple Sclerosis Society (National MS Society)
- SPP 2395, SCHI 1330/2-1, 4-1, and 11-1/Deutsche Forschungsgemeinschaft (German Research Foundation)
- ERC StG, No 950584/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
- SPP 2395/Deutsche Forschungsgemeinschaft (German Research Foundation)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
