

Best practices for germline variant and DNA methylation analysis of second- and third-generation sequencing data
- PMID: 39501379
- PMCID: PMC11536923
- DOI: 10.1186/s40246-024-00684-8
Best practices for germline variant and DNA methylation analysis of second- and third-generation sequencing data
Abstract
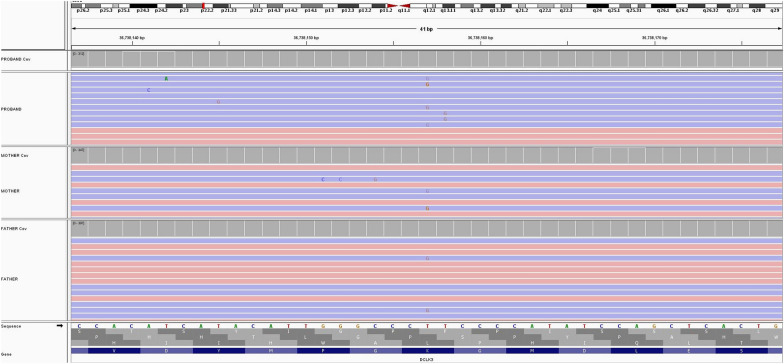
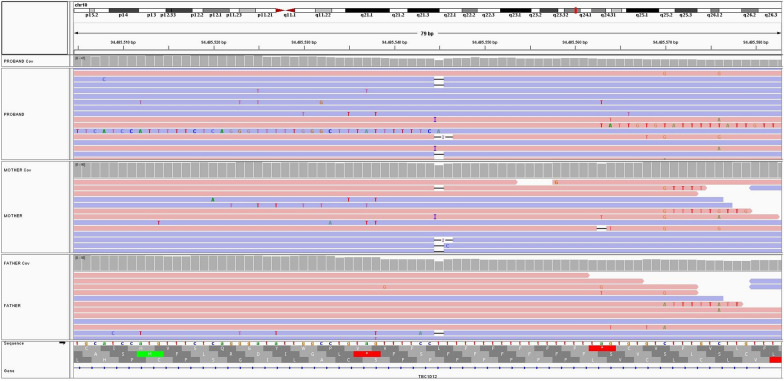
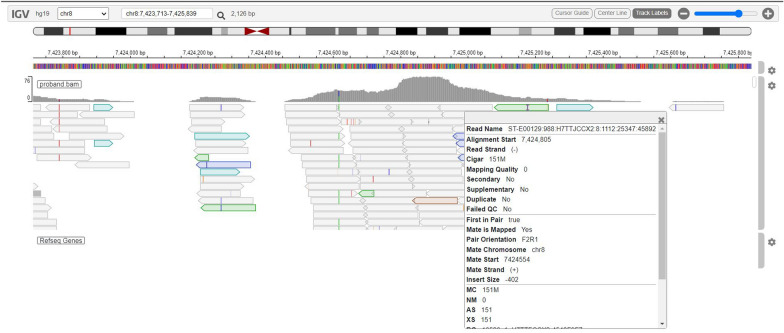
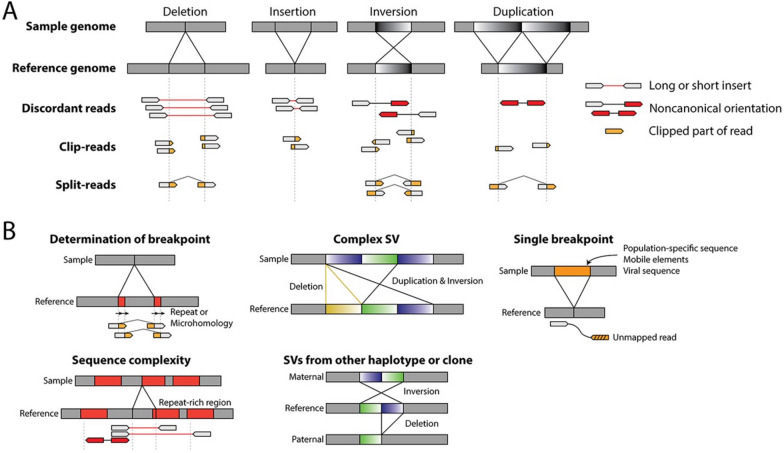
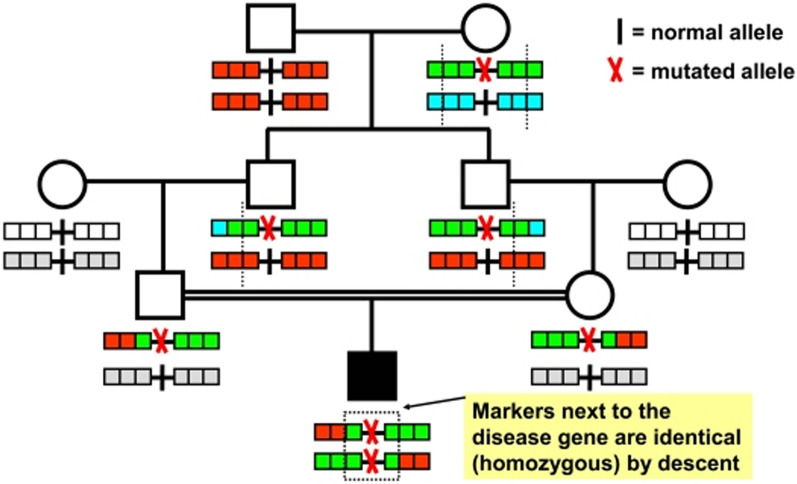
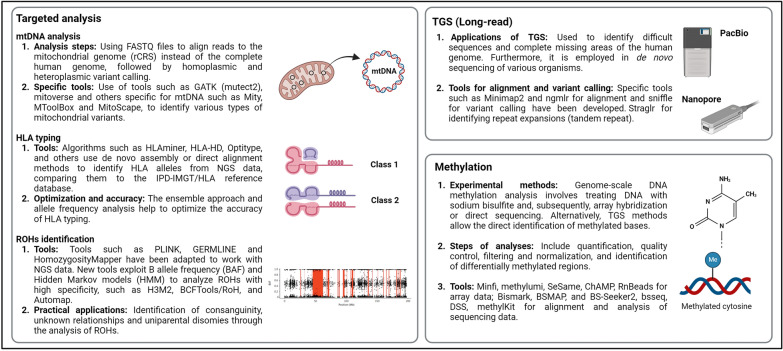
This comprehensive review provides insights and suggested strategies for the analysis of germline variants using second- and third-generation sequencing technologies (SGS and TGS). It addresses the critical stages of data processing, starting from alignment and preprocessing to quality control, variant calling, and the removal of artifacts. The document emphasized the importance of meticulous data handling, highlighting advanced methodologies for annotating variants and identifying structural variations and methylated DNA sites. Special attention is given to the inspection of problematic variants, a step that is crucial for ensuring the accuracy of the analysis, particularly in clinical settings where genetic diagnostics can inform patient care. Additionally, the document covers the use of various bioinformatics tools and software that enhance the precision and reliability of these analyses. It outlines best practices for the annotation of variants, including considerations for problematic genetic alterations such as those in the human leukocyte antigen region, runs of homozygosity, and mitochondrial DNA alterations. The document also explores the complexities associated with identifying structural variants and copy number variations, underscoring the challenges posed by these large-scale genomic alterations. The objective is to offer a comprehensive framework for researchers and clinicians, ensuring that genetic analyses conducted with SGS and TGS are both accurate and reproducible. By following these best practices, the document aims to increase the diagnostic accuracy for hereditary diseases, facilitating early diagnosis, prevention, and personalized treatment strategies. This review serves as a valuable resource for both novices and experts in the field, providing insights into the latest advancements and methodologies in genetic analysis. It also aims to encourage the adoption of these practices in diverse research and clinical contexts, promoting consistency and reliability across studies.
Keywords: Bioinformatics; DNA methylation; Genetic diagnostics; Germline variants; Hereditary diseases; NGS.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Hu T, Chitnis N, Monos D, Dinh A. Next-generation sequencing technologies: an overview. Hum Immunol. 2021;82:801–11. - PubMed
-
- Johnson S, Lee K, Riccitelli N. A comparison of Illumina and Element Biosciences sequencing platforms. Cancer Res. 2024;327(6_Supplement):327.
-
- Kumar KR, Cowley MJ, Davis RL. Next-generation sequencing and emerging technologies. Semin Thromb Hemost. 2019;45:661–73. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
