

Replication stress induces POLQ-mediated structural variant formation throughout common fragile sites after entry into mitosis
- PMID: 39505880
- PMCID: PMC11541566
- DOI: 10.1038/s41467-024-53917-8
Replication stress induces POLQ-mediated structural variant formation throughout common fragile sites after entry into mitosis
Abstract
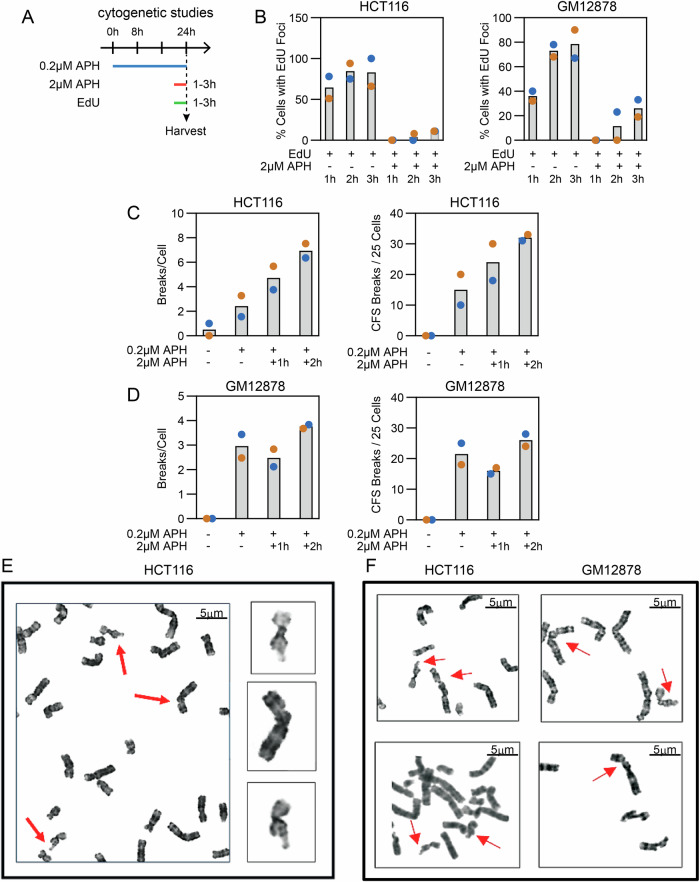
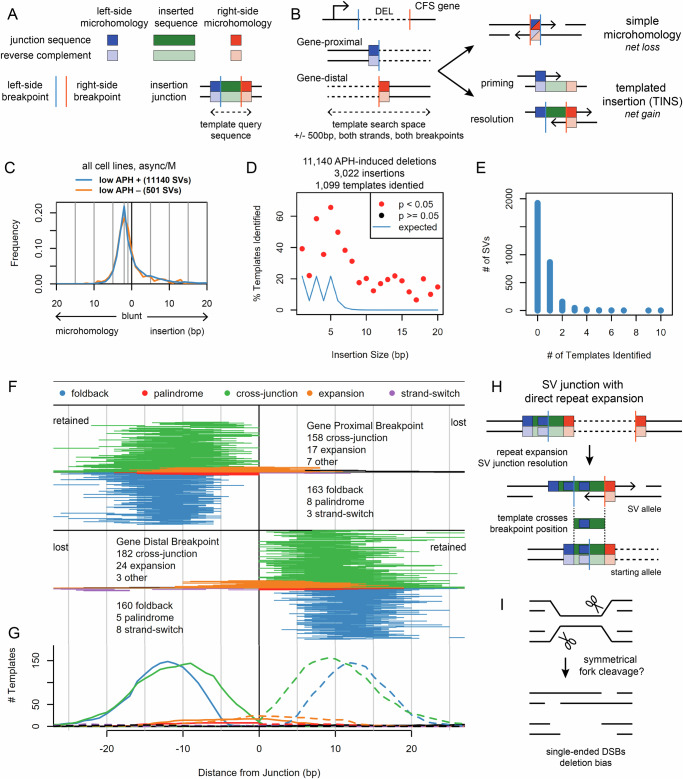
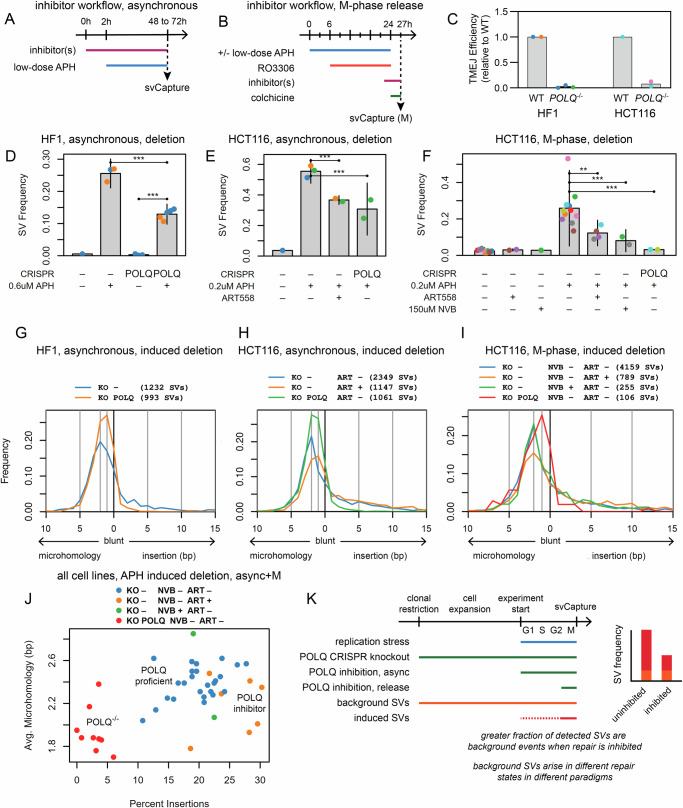
Genomic structural variants (SVs) greatly impact human health, but much is unknown about the mechanisms that generate the largest class of nonrecurrent alterations. Common fragile sites (CFSs) are unstable loci that provide a model for SV formation, especially large deletions, under replication stress. We study SV junction formation as it occurs in human cell lines by applying error-minimized capture sequencing to CFS DNA harvested after low-dose aphidicolin treatment. SV junctions form throughout CFS genes at a 5-fold higher rate after cells pass from G2 into M-phase. Neither SV formation nor CFS expression depend on mitotic DNA synthesis (MiDAS), an error-prone form of replication active at CFSs. Instead, analysis of tens of thousands of de novo SV junctions combined with DNA repair pathway inhibition reveal a primary role for DNA polymerase theta (POLQ)-mediated end-joining (TMEJ). We propose an important role for mitotic TMEJ in nonrecurrent SV formation genome wide.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- R01 CA200731/CA/NCI NIH HHS/United States
- R01 GM147026/GM/NIGMS NIH HHS/United States
- CA200731/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- GM147026/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
LinkOut - more resources
Full Text Sources
Research Materials
