

Enhancing transcription-replication conflict targets ecDNA-positive cancers
- PMID: 39506153
- PMCID: PMC11540844
- DOI: 10.1038/s41586-024-07802-5
Enhancing transcription-replication conflict targets ecDNA-positive cancers
Abstract
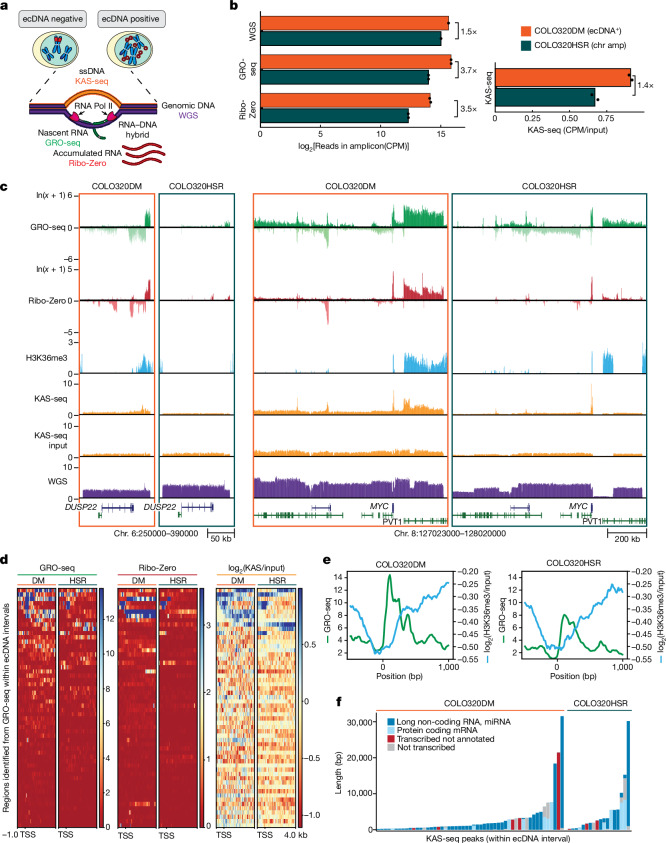
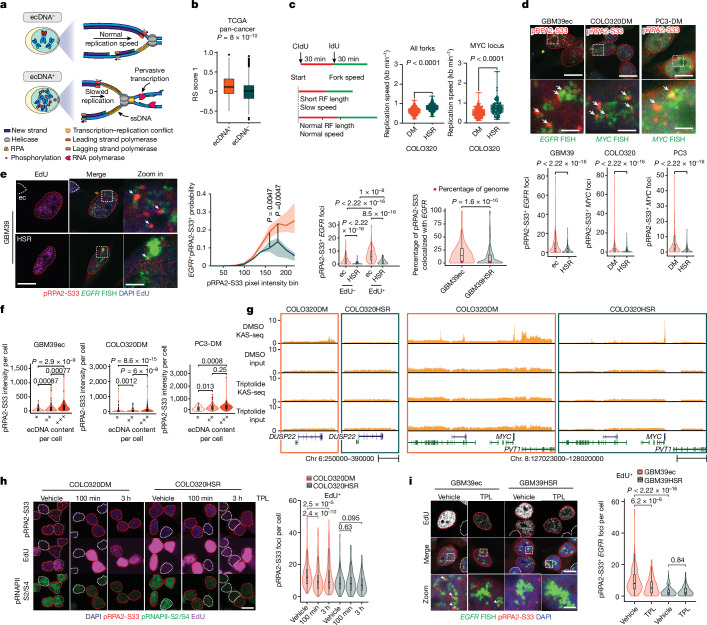
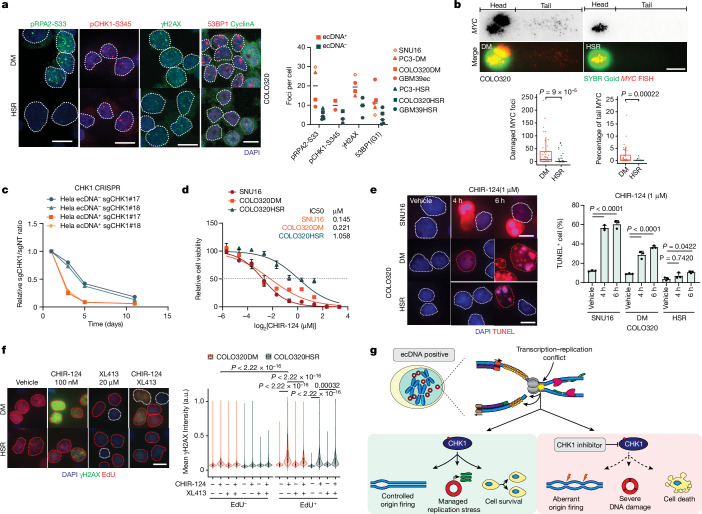
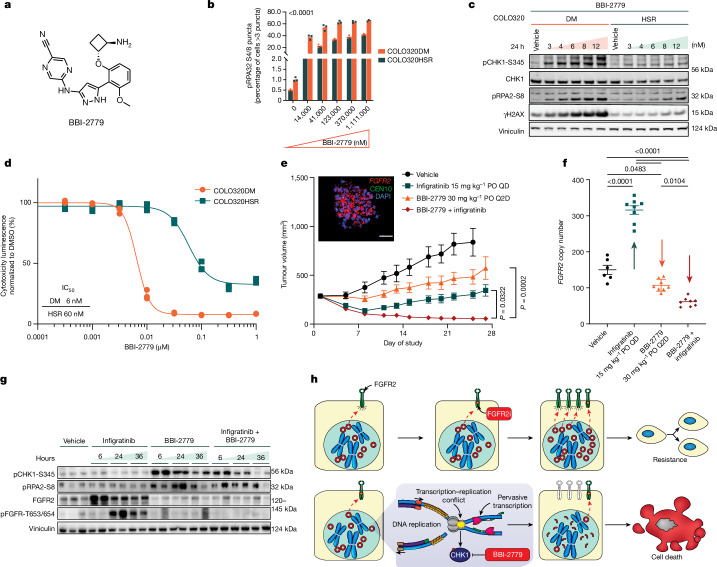
Extrachromosomal DNA (ecDNA) presents a major challenge for cancer patients. ecDNA renders tumours treatment resistant by facilitating massive oncogene transcription and rapid genome evolution, contributing to poor patient survival1-7. At present, there are no ecDNA-specific treatments. Here we show that enhancing transcription-replication conflict enables targeted elimination of ecDNA-containing cancers. Stepwise analyses of ecDNA transcription reveal pervasive RNA transcription and associated single-stranded DNA, leading to excessive transcription-replication conflicts and replication stress compared with chromosomal loci. Nucleotide incorporation on ecDNA is markedly slower, and replication stress is significantly higher in ecDNA-containing tumours regardless of cancer type or oncogene cargo. pRPA2-S33, a mediator of DNA damage repair that binds single-stranded DNA, shows elevated localization on ecDNA in a transcription-dependent manner, along with increased DNA double strand breaks, and activation of the S-phase checkpoint kinase, CHK1. Genetic or pharmacological CHK1 inhibition causes extensive and preferential tumour cell death in ecDNA-containing tumours. We advance a highly selective, potent and bioavailable oral CHK1 inhibitor, BBI-2779, that preferentially kills ecDNA-containing tumour cells. In a gastric cancer model containing FGFR2 amplified on ecDNA, BBI-2779 suppresses tumour growth and prevents ecDNA-mediated acquired resistance to the pan-FGFR inhibitor infigratinib, resulting in potent and sustained tumour regression in mice. Transcription-replication conflict emerges as a target for ecDNA-directed therapy, exploiting a synthetic lethality of excess to treat cancer.
© 2024. The Author(s).
Conflict of interest statement
H.Y.C., P.S.M. and V.B. are each a co-founder, advisor and have an equity interest in Boundless Bio. H.Y.C. is also a co-founder of Accent Therapeutics, Cartography Biosciences and Orbital Therapeutics, and an advisor of 10x Genomics, Arsenal Biosciences, Chroma Medicine, Exai Bio and Spring Discovery. S.J.B. is also an advisor and has an equity interest in Boundless Bio. S.C., E.T., S.G.M., R.H., J.P., A.S., S.M., S.T.M., A.B.P., S.K. and C.A.H. are employees of Boundless Bio. V.B. is a co-founder, consultant, SAB member and has equity interest in Abterra and Boundless Bio, and the terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict-of-interest policies. J.L. previously provided consulting services to Boundless Bio. The remaining authors declare no competing interests.
Figures










References
-
- Levan, A. & Levan, G. Have double minutes functioning centromeres? Hereditas88, 81–92 (1978). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
