

Identification of novel 3D-genome altering and complex structural variants underlying retinitis pigmentosa type 17 through a multistep and high-throughput approach
- PMID: 39507620
- PMCID: PMC11537883
- DOI: 10.3389/fgene.2024.1469686
Identification of novel 3D-genome altering and complex structural variants underlying retinitis pigmentosa type 17 through a multistep and high-throughput approach
Abstract
Introduction: Autosomal dominant retinitis pigmentosa type 17 (adRP, type RP17) is caused by complex structural variants (SVs) affecting a locus on chromosome 17 (chr17q22). The SVs disrupt the 3D regulatory landscape by altering the topologically associating domain (TAD) structure of the locus, creating novel TAD structures (neo-TADs) and ectopic enhancer-gene contacts. Currently, screening for RP17-associated SVs is not included in routine diagnostics given the complexity of the variants and a lack of cost-effective detection methods. The aim of this study was to accurately detect novel RP17-SVs by establishing a systematic and efficient workflow.
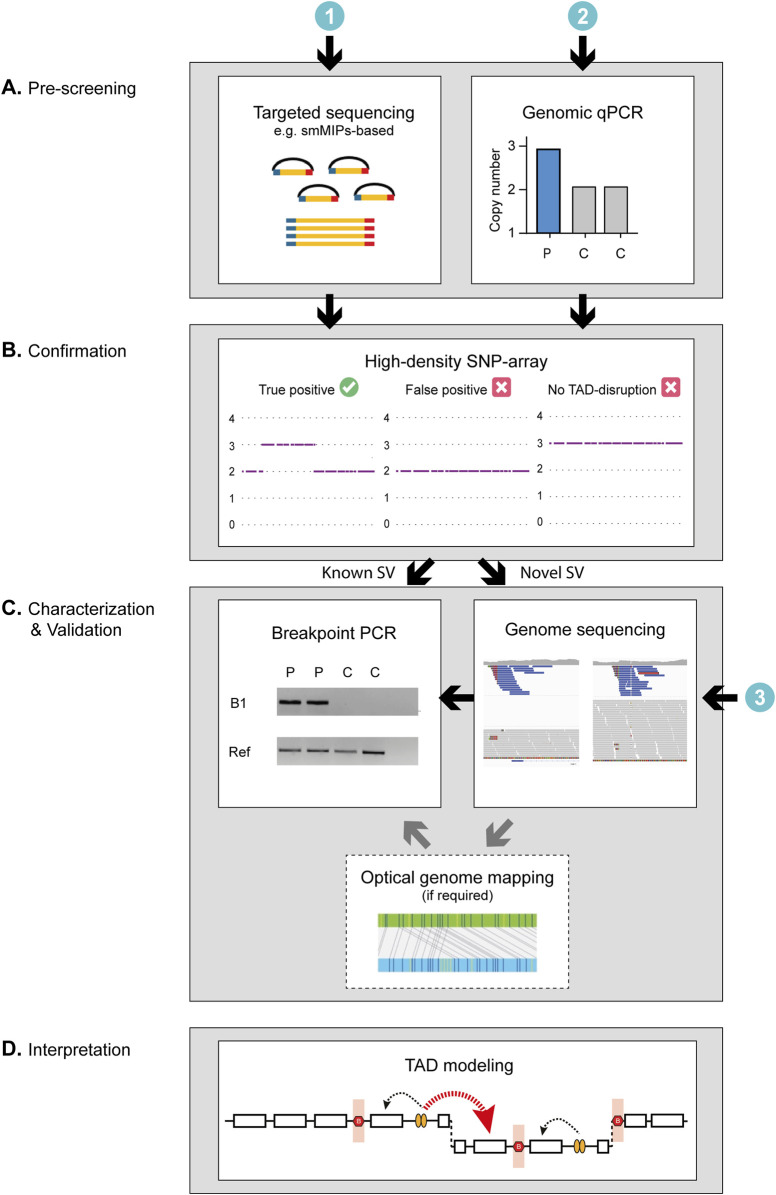
Methods: Genetically unexplained probands diagnosed with adRP (n = 509) from an international cohort were screened using a smMIPs or genomic qPCR-based approach tailored for the RP17 locus. Suspected copy number changes were validated using high-density SNP-array genotyping, and SV breakpoint characterization was performed by mutation-specific breakpoint PCR, genome sequencing and, if required, optical genome mapping. In silico modeling of novel SVs was performed to predict the formation of neo-TADs and whether ectopic contacts between the retinal enhancers and the GDPD1-promoter could be formed.
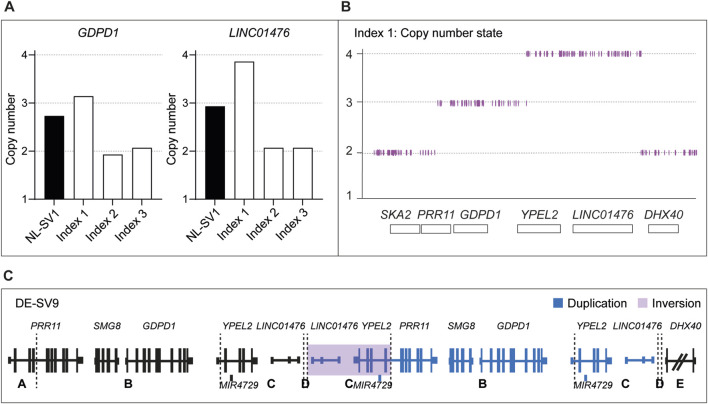
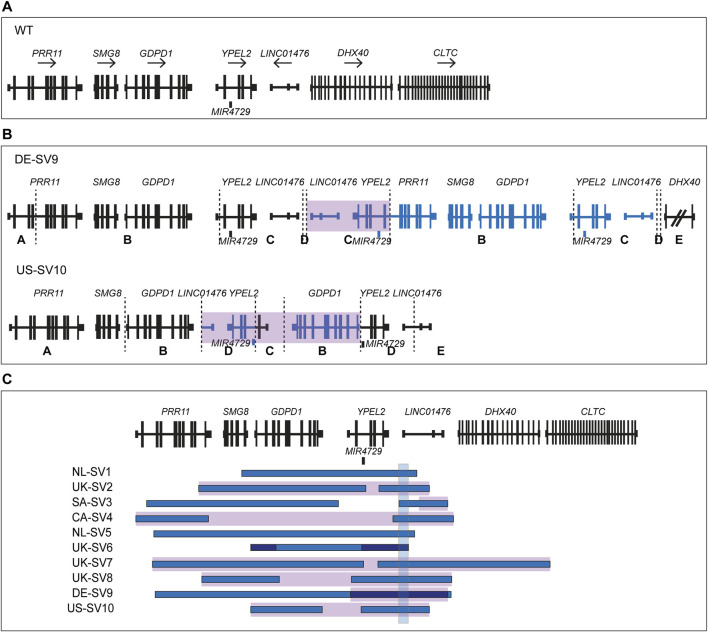
Results: Using this workflow, potential RP17-SVs were detected in eight probands of which seven were confirmed. Two novel SVs were identified that are predicted to cause TAD rearrangement and retinal enhancer-GDPD1 contact, one from Germany (DE-SV9) and three with the same SV from the United States (US-SV10). Previously reported RP17-SVs were also identified in three Australian probands, one with UK-SV2 and two with SA-SV3.
Discussion: In summary, we describe a validated multi-step pipeline for reliable and efficient RP17-SV discovery and expand the range of disease-associated SVs. Based on these data, RP17-SVs can be considered a frequent cause of adRP which warrants the inclusion of RP17-screening as a standard diagnostic test for this disease.
Keywords: gene diagnostics; gene regulation; inherited retinal dystrophies; retinitis pigmentosa; structural variants.
Copyright © 2024 de Bruijn, Panneman, Weisschuh, Cadena, Boonen, Holtes, Astuti, Cremers, Leijsten, Corominas, Gilissen, Skowronska, Woodley, Beggs, Toulis, Chen, Cheetham, Hardcastle, McLaren, Lamey, Thompson, Chen, de Roach, Urwin, Sullivan, Roosing.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
-
- de Bruijn S. E., Fiorentino A., Ottaviani D., Fanucchi S., Melo U. S., Corral-Serrano J. C., et al. (2020). Structural variants create new topological-associated domains and ectopic retinal enhancer-gene contact in dominant retinitis pigmentosa. Am. J. Hum. Genet. 107 (5), 802–814. 10.1016/j.ajhg.2020.09.002 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Research Materials