

Microbial occurrence and symbiont detection in a global sample of lichen metagenomes
- PMID: 39509454
- PMCID: PMC11542873
- DOI: 10.1371/journal.pbio.3002862
Microbial occurrence and symbiont detection in a global sample of lichen metagenomes
Abstract
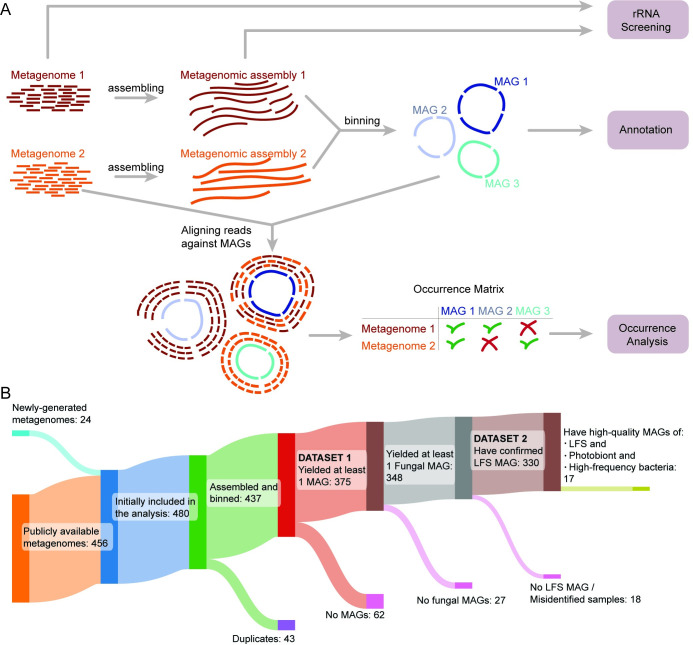
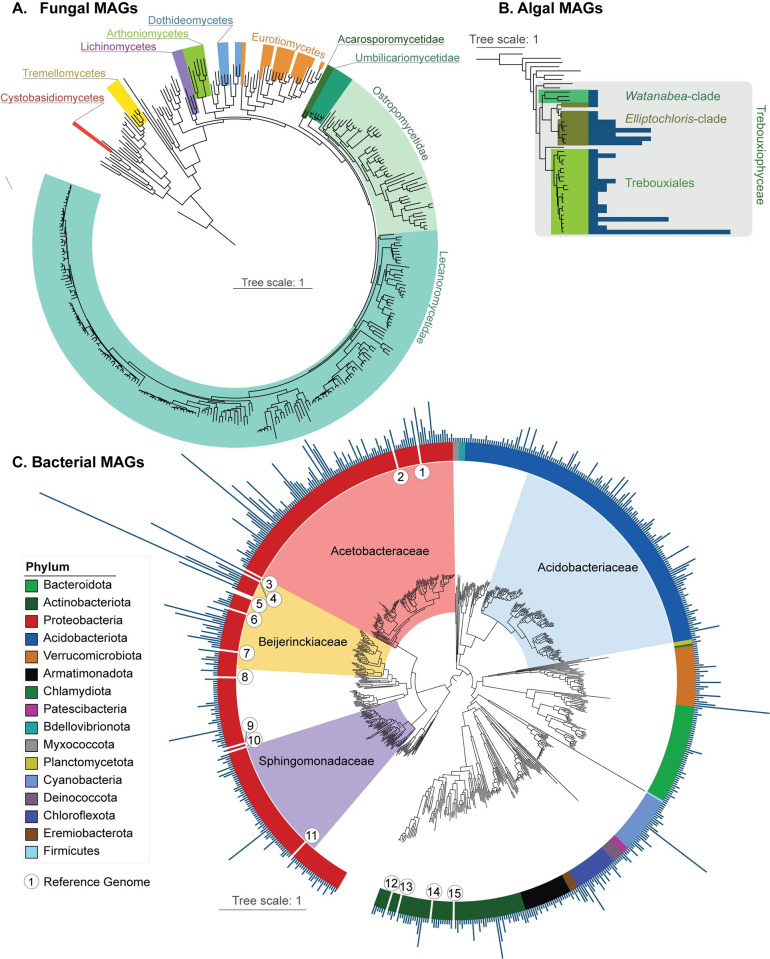
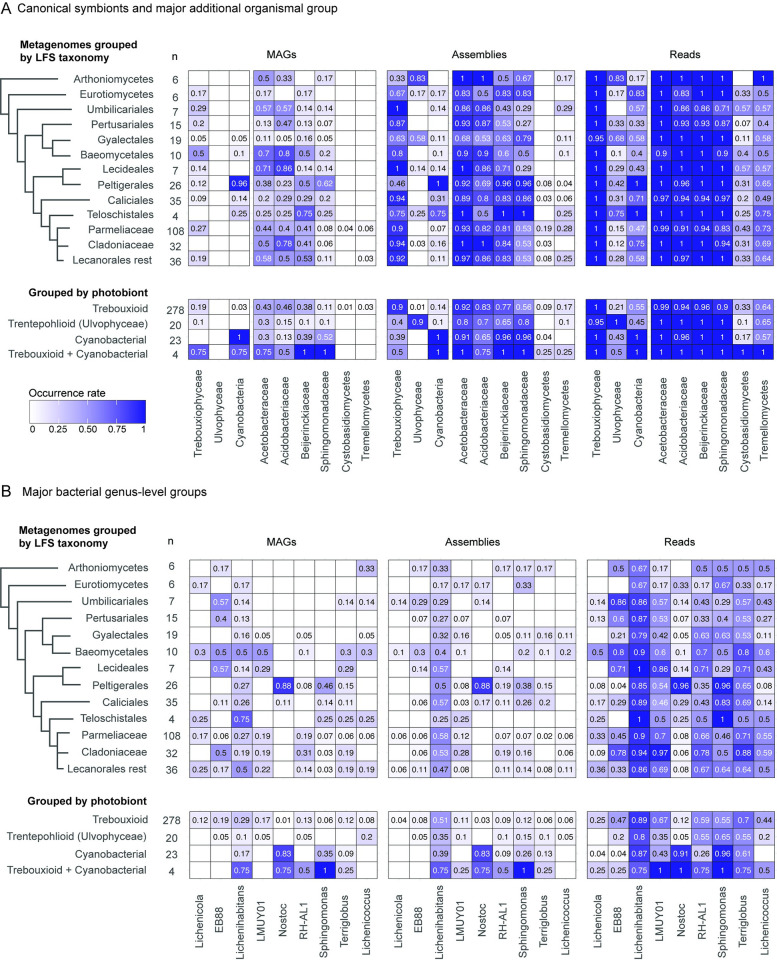
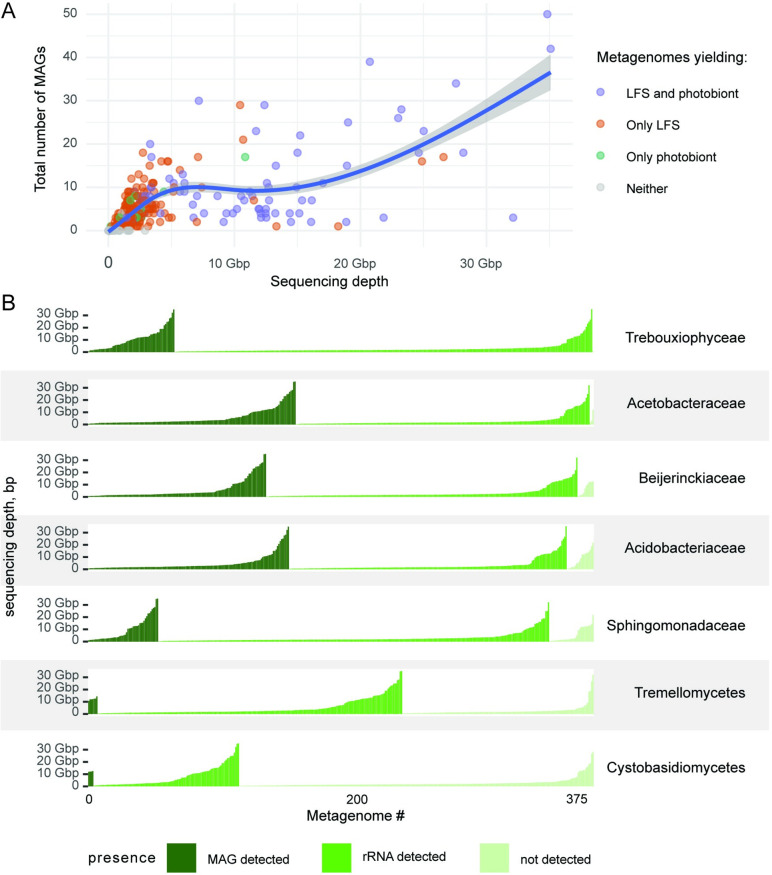
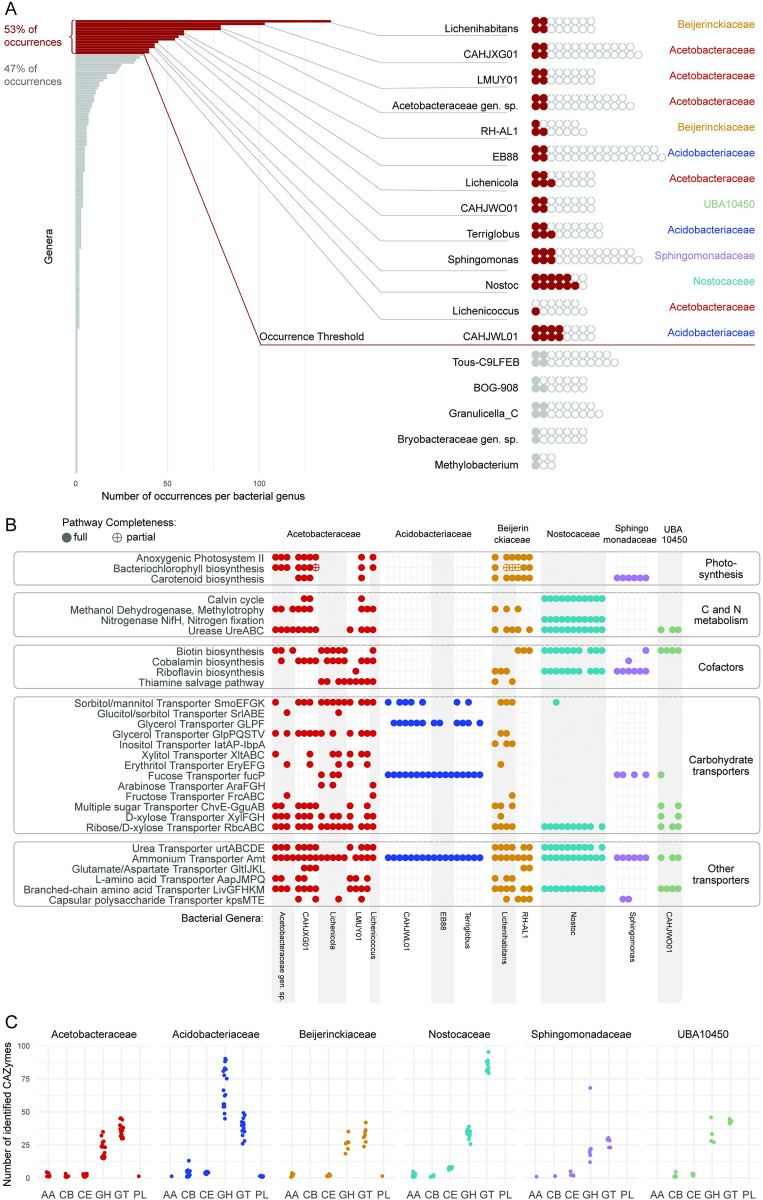
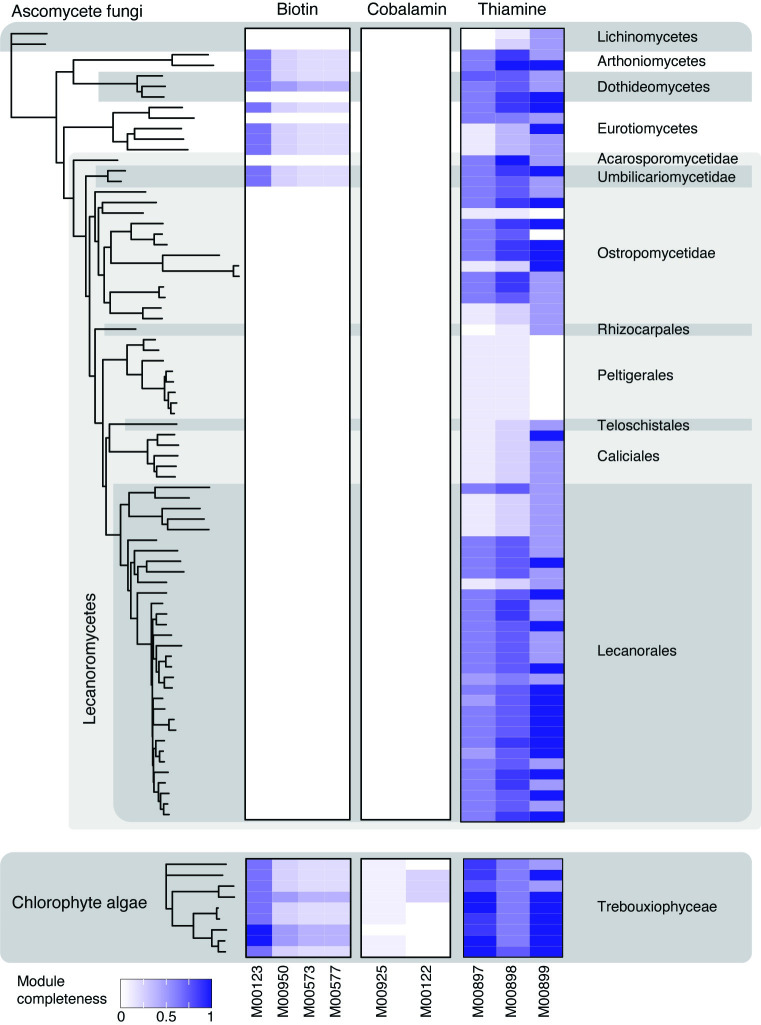
In lichen research, metagenomes are increasingly being used for evaluating symbiont composition and metabolic potential, but the overall content and limitations of these metagenomes have not been assessed. We reassembled over 400 publicly available metagenomes, generated metagenome-assembled genomes (MAGs), constructed phylogenomic trees, and mapped MAG occurrence and frequency across the data set. Ninety-seven percent of the 1,000 recovered MAGs were bacterial or the fungal symbiont that provides most cellular mass. Our mapping of recovered MAGs provides the most detailed survey to date of bacteria in lichens and shows that 4 family-level lineages from 2 phyla accounted for as many bacterial occurrences in lichens as all other 71 families from 16 phyla combined. Annotation of highly complete bacterial, fungal, and algal MAGs reveals functional profiles that suggest interdigitated vitamin prototrophies and auxotrophies, with most lichen fungi auxotrophic for biotin, most bacteria auxotrophic for thiamine and the few annotated algae with partial or complete pathways for both, suggesting a novel dimension of microbial cross-feeding in lichen symbioses. Contrary to longstanding hypotheses, we found no annotations consistent with nitrogen fixation in bacteria other than known cyanobacterial symbionts. Core lichen symbionts such as algae were recovered as MAGs in only a fraction of the lichen symbioses in which they are known to occur. However, the presence of these and other microbes could be detected at high frequency using small subunit rRNA analysis, including in many lichens in which they are not otherwise recognized to occur. The rate of MAG recovery correlates with sequencing depth, but is almost certainly influenced by biological attributes of organisms that affect the likelihood of DNA extraction, sequencing and successful assembly, including cellular abundance, ploidy and strain co-occurrence. Our results suggest that, though metagenomes are a powerful tool for surveying microbial occurrence, they are of limited use in assessing absence, and their interpretation should be guided by an awareness of the interacting effects of microbial community complexity and sequencing depth.
Copyright: © 2024 Tagirdzhanova et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
-
- Schwendener S. Die Algentypen der Flechtengonidien. Basel: Universitaetsbuchdruckerei; 1869.
-
- De Bary A. Die Erscheinung der Symbiose. Strassburg: Verlag Karl Trübner; 1879.
-
- Drew EA, Smith DC. Studies in the physiology of lichens: VIII. Movement of glucose from alga to fungus during photosynthesis in the thallus of Peltigera polydactyla. New Phytol. 1967. Jul;66(3):389–400.
-
- Ahmadjian V. The lichen symbiosis. New York: John Wiley & Sons; 1993.
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
