

SNX9 family mediates βarrestin-independent GPCR endocytosis
- PMID: 39511325
- PMCID: PMC11544122
- DOI: 10.1038/s42003-024-07157-7
SNX9 family mediates βarrestin-independent GPCR endocytosis
Abstract
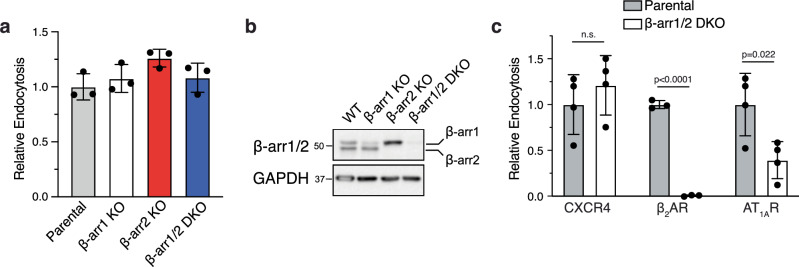
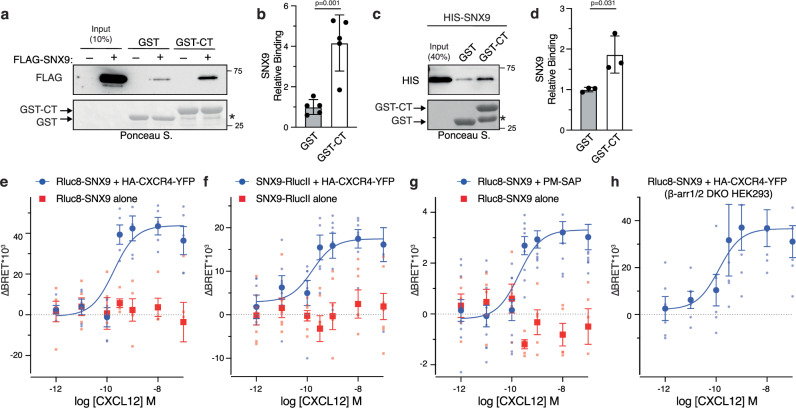
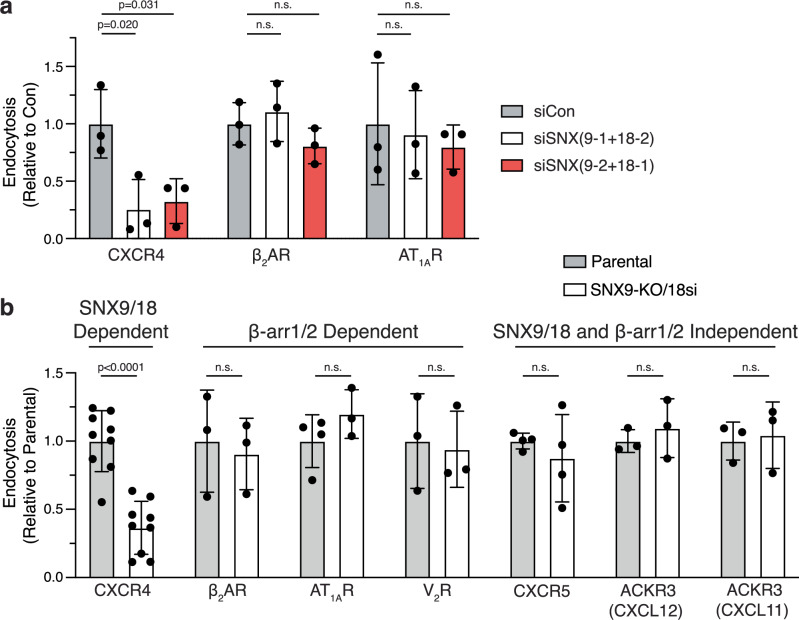
Agonist-stimulated GPCR endocytosis typically occurs via the multi-faceted adaptor proteins known as βarrestins. However, endocytosis of several GPCRs occurs independently of β-arrestins, suggesting an additional mode of GPCR endocytosis, but the mechanisms remain unknown. Here we provide evidence that sorting nexin 9 (SNX9), a previously described endocytic remodeling protein, functions as a novel cargo adaptor that promotes agonist-stimulated GPCR endocytosis. We show that SNX9 and SNX18, but not β-arrestins, are necessary for endocytosis of the chemokine receptor CXCR4. SNX9 is recruited to CXCR4 at the plasma membrane and interacts directly with the carboxyl-terminal tail of the receptor in a phosphorylation-dependent manner. We also provide evidence that some receptors do not require SNX9 and SNX18 nor β-arrestins for endocytosis, suggesting additional modes for GPCR endocytosis. These results provide novel insights into the mechanisms regulating GPCR trafficking and broaden our overall understanding of GPCR regulation.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
