

Outgrowth of Escherichia is susceptible to aggravation of systemic lupus erythematosus
- PMID: 39511594
- PMCID: PMC11542361
- DOI: 10.1186/s13075-024-03413-7
Outgrowth of Escherichia is susceptible to aggravation of systemic lupus erythematosus
Abstract
Background: Systemic lupus erythematosus (SLE) is linked to host gut dysbiosis. Here we performed faecal gut microbiome sequencing to investigate SLE-pathogenic gut microbes and their potential mechanisms.
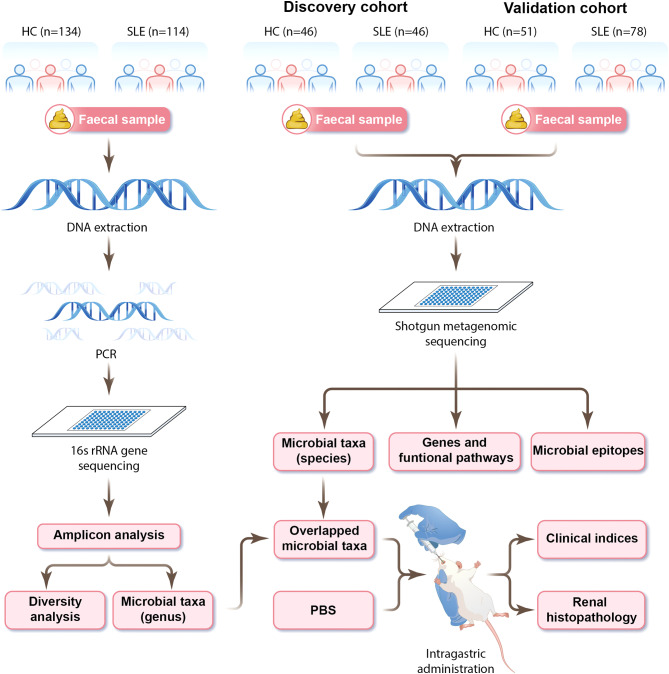
Methods: There were 134 healthy controls (HCs) and 114 SLE cases for 16 S ribosomal RNA (rRNA) sequencing and 97 HCs and 124 SLE cases for shotgun metagenomics. Faecal microbial changes and associations with clinical phenotypes were evaluated, and SLE-associated microbial genera were identified in amplicon analysis. Next, metagenomic sequencing was applied for accurate identification of microbial species and discovery of their metabolic pathways and immunogenic peptides both relevant to SLE. Finally, contribution of specific taxa to disease development was confirmed by oral gavage into lupus-prone MRL/lpr mice.
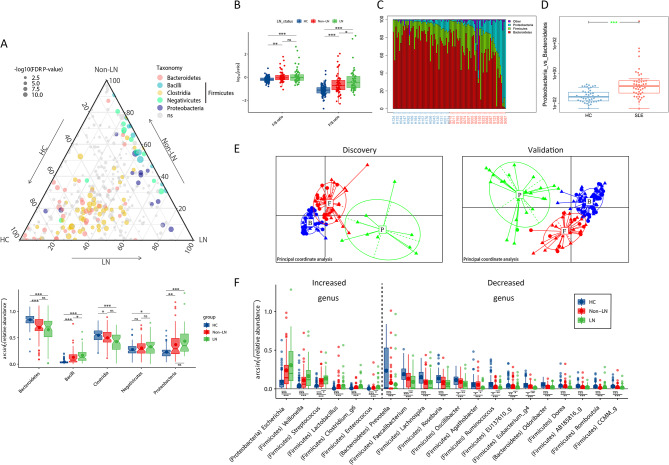
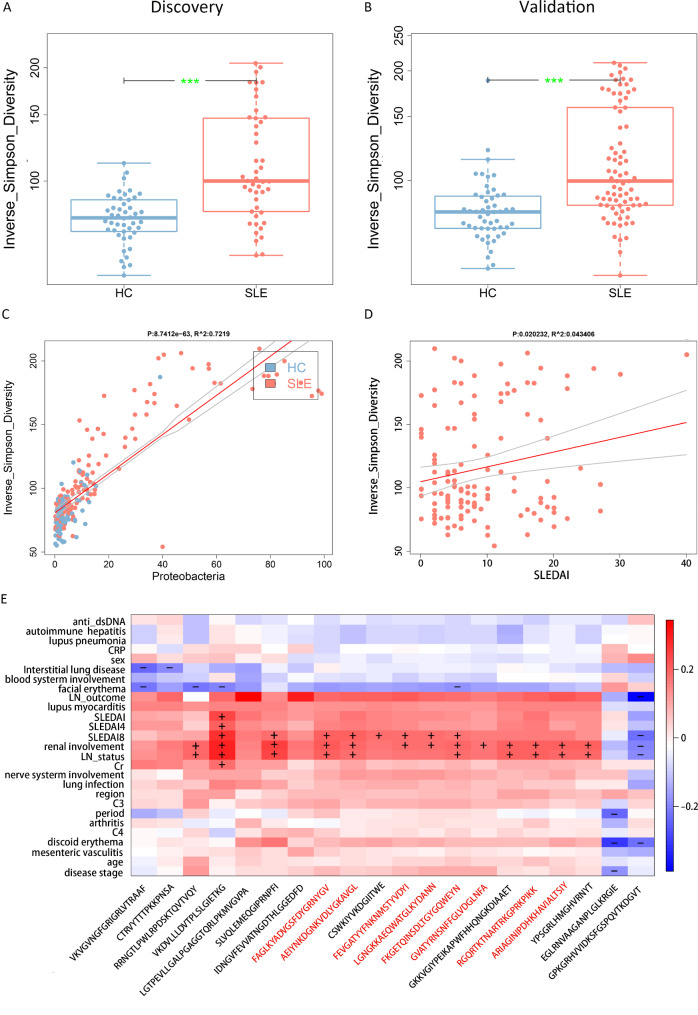
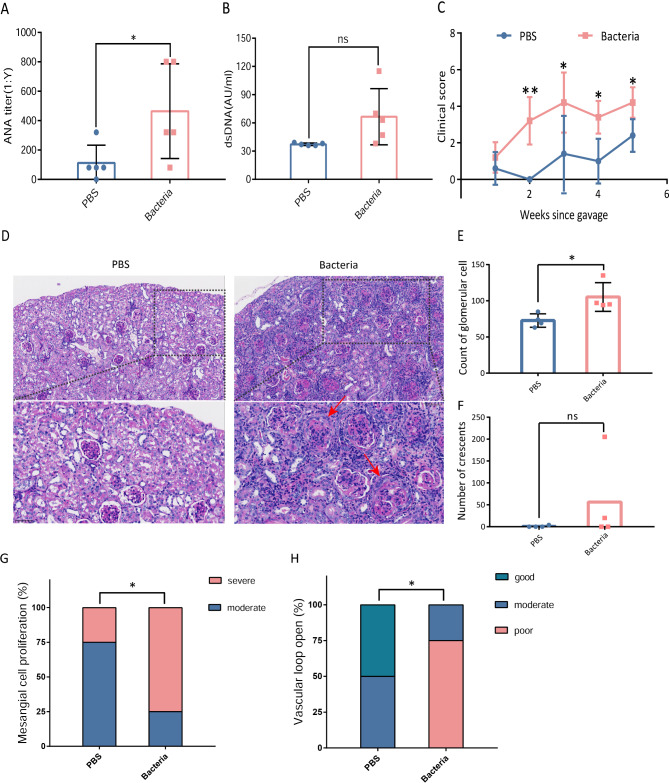
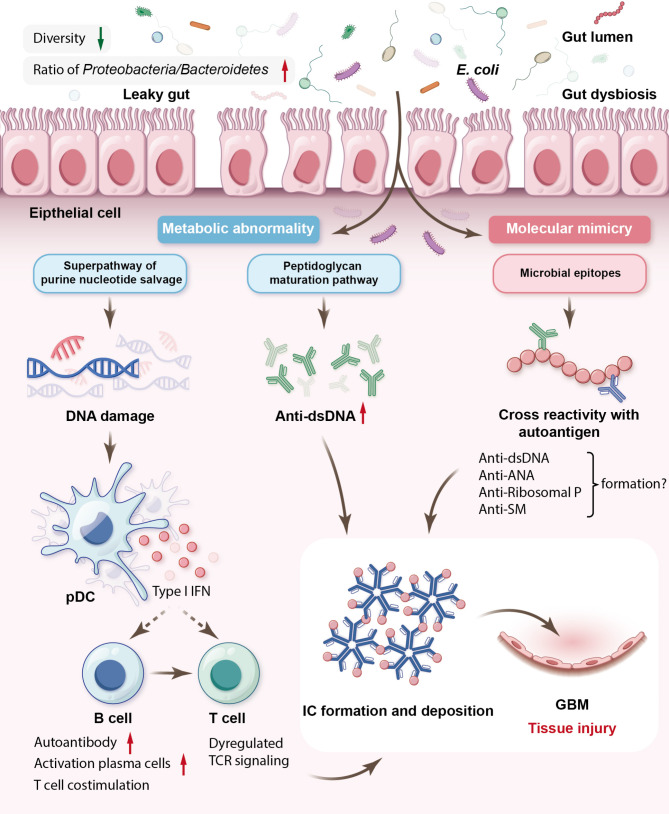
Results: SLE patients had gut microbiota richness reduction and composition alteration, particularly lupus nephritis and active patients. Proteobacteria/Bacteroidetes (P/B) ratio was remarkably up-regulated, and Escherichia was identified as the dominantly expanded genus in SLE, followed by metagenomics accurately located Escherichia coli and Escherichia unclassified species. Significant associations primarily appeared among Escherichia coli, metabolic pathways of purine nucleotide salvage or peptidoglycan maturation and SLE disease activity index (SLEDAI), and between multiple epitopes from Escherichia coli and disease activity or renal involvement phenotype. Finally, gavage with faecal Escherichia revealed that it upregulated lupus-associated serum traits and aggravated glomerular lesions in MRL/lpr mice.
Conclusion: We characterize a novel SLE exacerbating Escherichia outgrowth and suggest its contribution to SLE procession may be partially associated with metabolite changes and cross-reactivity of gut microbiota-associated epitopes and host autoantigens. The findings could provide a deeper insight into gut Escherichia in the procession of SLE.
Keywords: Escherichia; Gut microbiota dysbiosis; Gut microbiota-associated epitopes; Shotgun metagenomics; Systemic lupus erythematosus.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. 2016;2:16039. - PubMed
-
- Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365:2110–21. - PubMed
-
- Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13(5):321–35. - PubMed
-
- Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. 2015;21(8):895–905. - PubMed
MeSH terms
Grants and funding
- 2024A1515010590/Natural Science Foundation of Guangdong Province, China
- JCYJ20220530154601004/Shenzhen Science and Technology Innovation Bureau
- 2020B1111170008/Guangdong Clinical Research Center of Immune disease
- SW201901/Ten & Five Project of the Third Affiliated Hospital of Sun Yat-Sen University
- A2675/Distinguished Young Scholar Candidates Program for The Third Affiliated Hospital of Sun Yat-Sen University
LinkOut - more resources
Full Text Sources
Medical
