

Cytometry masked autoencoder: An accurate and interpretable automated immunophenotyper
- PMID: 39515318
- PMCID: PMC11604491
- DOI: 10.1016/j.xcrm.2024.101808
Cytometry masked autoencoder: An accurate and interpretable automated immunophenotyper
Abstract
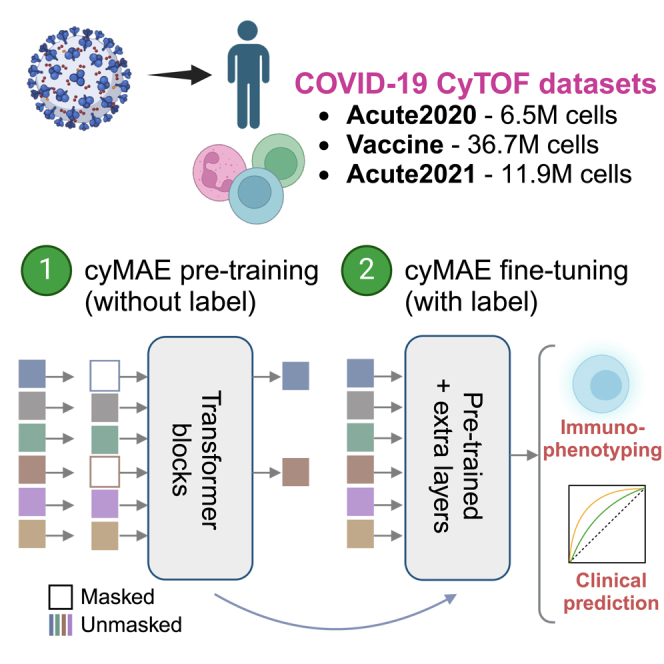
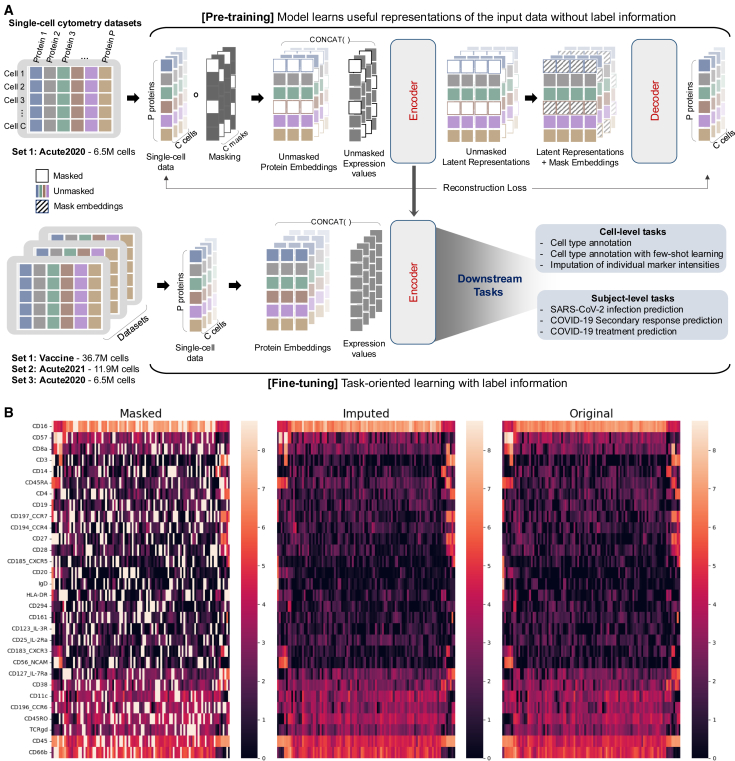
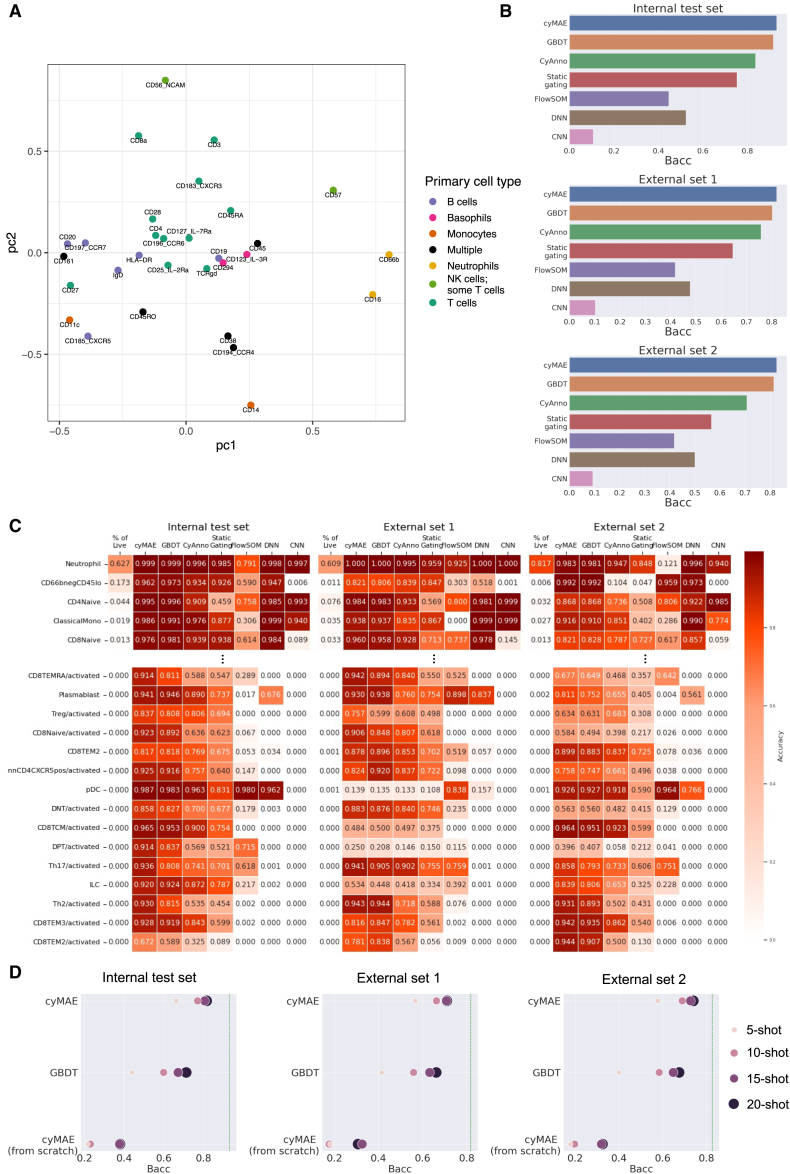
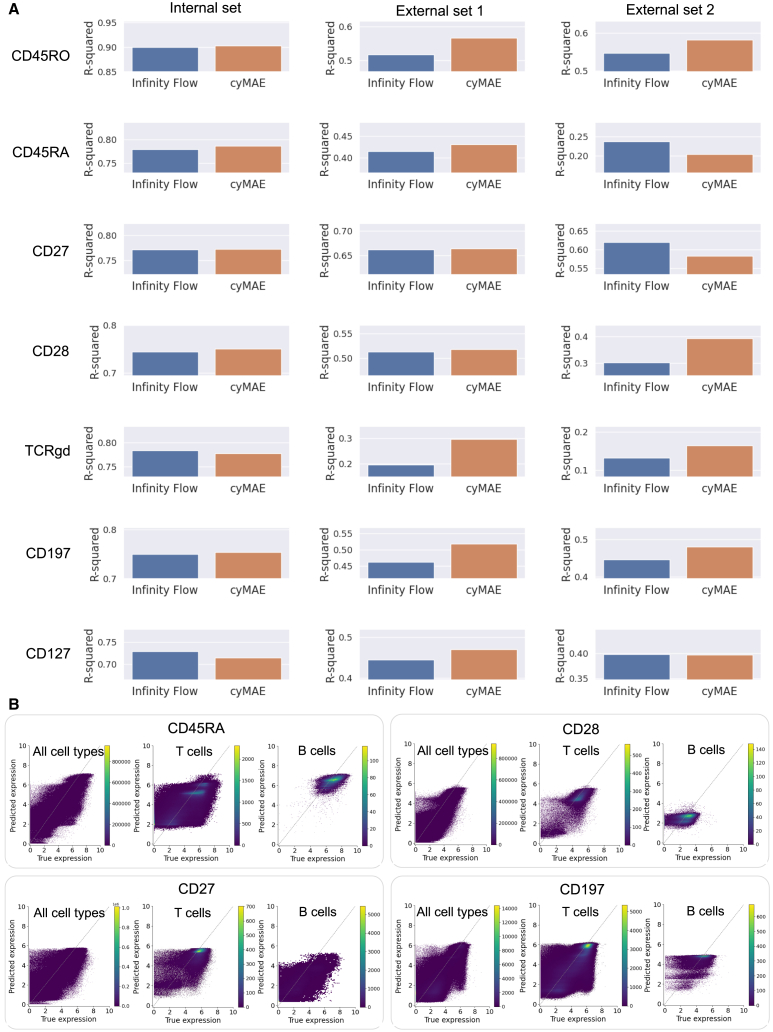
Single-cell cytometry data are crucial for understanding the role of the immune system in diseases and responses to treatment. However, traditional methods for annotating cytometry data face challenges in scalability, robustness, and accuracy. We propose a cytometry masked autoencoder (cyMAE), which automates immunophenotyping tasks including cell type annotation. The model upholds user-defined cell type definitions, facilitating interpretability and cross-study comparisons. The training of cyMAE has a self-supervised phase, which leverages large amounts of unlabeled data, followed by fine-tuning on specialized tasks using smaller amounts of annotated data. The cost of training a new model is amortized over repeated inferences on new datasets using the same panel. Through validation across multiple studies using the same panel, we demonstrate that cyMAE delivers accurate and interpretable cellular immunophenotyping and improves the prediction of subject-level metadata. This proof of concept marks a significant step forward for large-scale immunology studies.
Keywords: automated gating; deep learning; high-dimensional cytometry; immunophenotyping; machine learning; mass cytometry; representation learning.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests E.J.W. is a member of the Parker Institute for Cancer Immunotherapy, which supports cancer immunotherapy research in his laboratory. E.J.W. is an advisor for Arsenal Biosciences, Coherus, Danger Bio, IpiNovyx, NewLimit, Marengo, Pluto Immunotherapeutics, Related Sciences, Santa Ana Bio, and Synthekine. E.J.W. is a founder of and holds stock in Coherus, Danger Bio, Prox Biosciences, and Arsenal Biosciences.
Figures
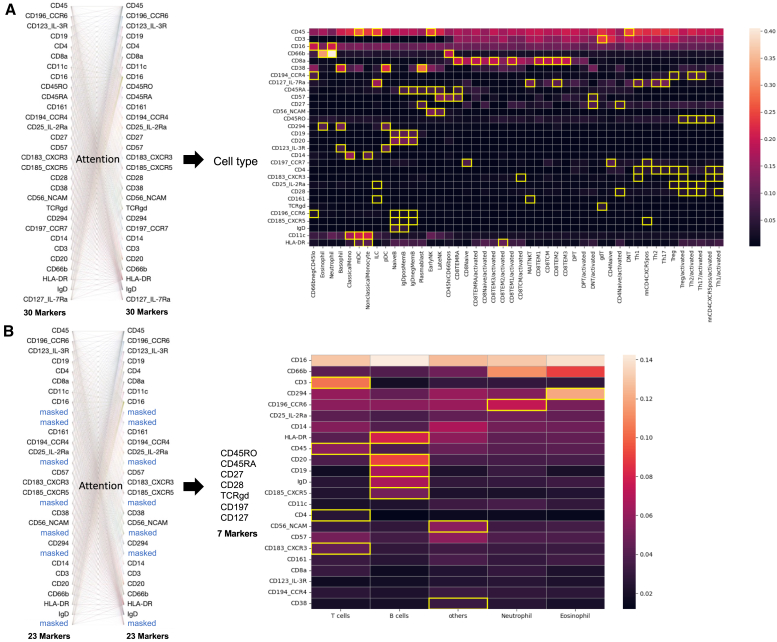
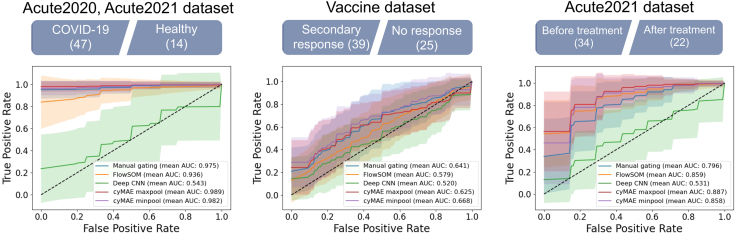
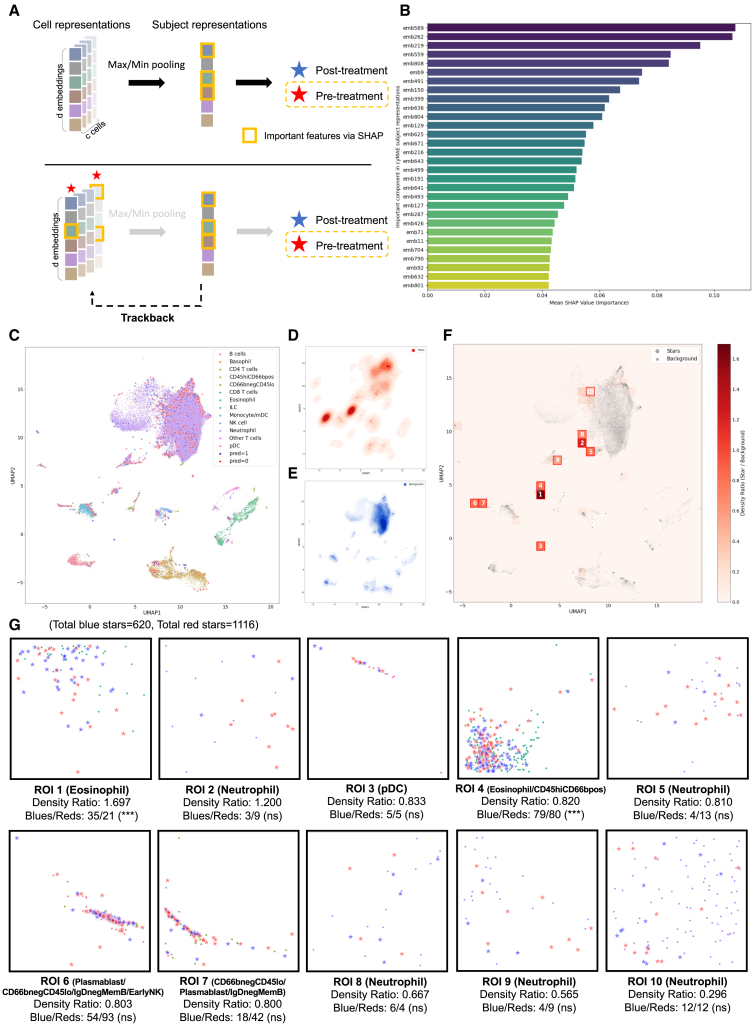
References
-
- Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir E.a.D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R., et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
